En biología evolutiva, los caracteres de los organismos pueden representarse como rasgos discretos (como tipo de dieta, hábitat o sistema de apareamiento) o continuos (como tamaño corporal, longitud del ala o temperatura óptima). Analizar cómo estos caracteres han cambiado a lo largo del tiempo nos permite inferir los procesos que han moldeado la diversidad biológica actual.
La reconstrucción de estados ancestrales es una herramienta fundamental para estimar los valores o estados que tenían estos caracteres en los ancestros comunes de las especies actuales. Para ello, utilizamos modelos estadísticos que describen cómo evolucionan los rasgos sobre un árbol filogenético.
Modelos de evolución de caracteres continuos
Los modelos que exploraremos pertenecen a la familia de modelos gaussianos, es decir, aquellos que suponen que los cambios evolutivos siguen una distribución normal. En particular, nos enfocaremos en dos clases principales:
Modelos de Movimiento Browniano (BM): Suponen que el rasgo evoluciona como un paseo aleatorio, con una media de cambio igual a cero y una varianza proporcional al tiempo. Son útiles para estimar una tasa global de evolución y explorar cómo esta puede variar entre linajes o a lo largo del tiempo.
Modelos de Ornstein-Uhlenbeck (OU): Introducen un parámetro adicional que representa un valor óptimo hacia el cual tiende la evolución del carácter. Este tipo de modelo es más adecuado cuando se sospecha que la evolución del rasgo está sujeta a selección estabilizadora.
En este caso, utilizamos datos morfológicos provenientes del artículo Analyses of head and thorax in Eupomphini (Meloidae) suggest that complex behaviors are not associated to changes in general shape. La matriz de caracteres (med_geiger.nex) incluye cuatro medidas continuas obtenidas de especies de escarabajos del grupo Eupomphini: volumen de élitros, volumen del abdomen, amplitud y convexidad. Estas características se utilizarán para modelar su evolución a lo largo de un árbol filogenético calibrado en el tiempo, que corresponde a una versión modificada del árbol utilizado en la primera unidad sobre patrones de diversificación.
Descarga del Árbol Filogenético y la matriz de caracteres continuos
📥 Puedes descargar el archivo .tre con el árbol en el siguiente enlace:
Guarda los archivos en la carpeta correspondiente y verifica su ubicación antes de continuar.
Modelo Browniano de tasa constante en RevBayes
En modelos de evolución de caracteres continuos como el Movimiento Browniano, se asume que el carácter cambia de forma aleatoria a lo largo del tiempo, sin una dirección preferida. En este contexto, el parámetro \(\sigma^2\) representa la tasa de evolución del rasgo: a mayor valor de \(\sigma^2\), mayor es la varianza esperada en el cambio del carácter entre especies, lo que implica una evolución más rápida.
Al comparar la distribución a priori y posterior de \(\sigma^2\) mediante análisis bayesianos, podemos evaluar cuánto han informado los datos al modelo. Una diferencia clara entre ambas distribuciones indica que los datos aportan evidencia sobre la velocidad de evolución del carácter evaluado.
🧠 ¿Qué es \(\sigma^2\) en este contexto?
En un modelo de Movimiento Browniano aplicado a evolución de caracteres:
\[ Var(cambio-del-carácter) = \sigma^2 * t \]
t: longitud de la rama (tiempo evolutivo).
σ²: parámetro de tasa de evolución.
El cambio esperado del carácter es 0, pero la varianza esperada del cambio crece con el tiempo y con \(\sigma^2\).
📌 ¿Qué implica eso para tu análisis?
Cuando tú analizas, por ejemplo, el volumen del élitro, estás suponiendo que ese carácter cambia aleatoriamente en el tiempo, pero a un ritmo que depende de \(\sigma^2\). Entonces:
Un \(\sigma^2\) alto → el rasgo cambia mucho a lo largo del árbol → evolución rápida del carácter.
Un \(\sigma^2\) bajo → el rasgo cambia poco → evolución lenta o conservada.
En análisis bayesianos, queremos estimar la probabilidad de un parámetro (como \(\sigma^2\), la tasa de evolución bajo el modelo Browniano) después de ver los datos. Para eso, necesitamos comparar dos cosas:
1️⃣ El script de posterior (mcmc_BM.Rev)
Utiliza tanto el modelo como los datos observados.
Fija los datos con X.clamp(data).
Nos dice: 👉 ¿Qué valores de\(\sigma^2\) son más probables ahora que hemos visto los datos?
################################################################################# Ejemplo de RevBayes: Inferencia bayesiana de tasas de evolución bajo un # modelo de Movimiento Browniano con tasa constante############################################################################################################### Lectura de los datos ################################ Seleccionar el carácter continuo a analizar (ej. volumen del élitros)trait <-1# Leer el árbol filogenético calibrado en el tiempoT <-readTrees("../data/subarbol_ingroup_morph.nex")[1]# Leer la matriz de caracteres continuosdata <-readContinuousCharacterData("../data/med_geiger.nex")data.excludeAll()data.includeCharacter(trait)# Crear vectores para movimientos (moves) y monitoresmoves =VectorMoves()monitors =VectorMonitors()################################### Especificar el modelo del árbol ###################################tree <- T####################################### Especificar el modelo de la tasa σ² ######################################## Definir una distribución a priori log-uniforme para la tasa de evoluciónsigma2 ~dnLoguniform(1e-5, 1e-1)# Movimiento de escala para proponer nuevos valores de sigma2moves.append( mvScale(sigma2, weight=1.0) )####################################### Especificar el modelo BM (Browniano) ######################################## Modelo con REML y sigma2 como tasa de cambio (desviación estándar)X ~dnPhyloBrownianREML(tree, branchRates=sqrt(sigma2))# Fijar los datos observados al nodo estocásticoX.clamp(data)###################### Crear el modelo completo ######################mymodel =model(sigma2)# Configurar monitores para guardar resultados y mostrar en pantallamonitors.append( mnModel(filename="../out/morpho/simple_BM.log", printgen=10) )monitors.append( mnScreen(printgen=100, sigma2) )########################### Ejecutar la simulación MCMC ############################ Crear objeto MCMCmymcmc =mcmc(mymodel, monitors, moves, nruns=2, combine="mixed")# Ejecución principalmymcmc.run(generations=3000)# Finalizarq()
2️⃣ El script de prior (mcmc_BM_prior.Rev)
Usa solo el modelo y su distribución a priori, sin considerar los datos.
Omite los datos con mymodel.ignoreAllData().
Nos dice: 👉 ¿Qué creíamos posible sobre\(\sigma^2\) antes de ver los datos?
################################################################################# Ejemplo de RevBayes: Muestreo de la distribución a priori del modelo # de Movimiento Browniano con tasa constante############################################################################################################### Lectura de los datos ################################ Leer el árbol filogenético calibradoT <-readTrees("../data/subarbol_ingroup_morph.nex")[1]# Crear vectores para movimientos y monitoresmoves =VectorMoves()monitors =VectorMonitors()################################### Modelo del árbol ###################################tree <- T####################################### Modelo de tasa con distribución a priori #######################################sigma2 ~dnLoguniform(1e-5, 1e-1)moves.append( mvScale(sigma2, weight=1) )####################################### Modelo BM sin usar datos observados #######################################X ~dnPhyloBrownianREML(tree, branchRates=sqrt(sigma2))###################### Crear el modelo y omitir datos ######################mymodel =model(sigma2)mymodel.ignoreAllData()# Monitores para archivo y pantallamonitors.append( mnModel(filename="../out/morpho/simple_BM_prior.log", printgen=10) )monitors.append( mnScreen(printgen=100, sigma2) )########################### Ejecutar simulación MCMC ###########################mymcmc =mcmc(mymodel, monitors, moves, nruns=2, combine="mixed")# Solo ejecución, sin burn-in necesario para priormymcmc.run(generations=3000)# Finalizarq()
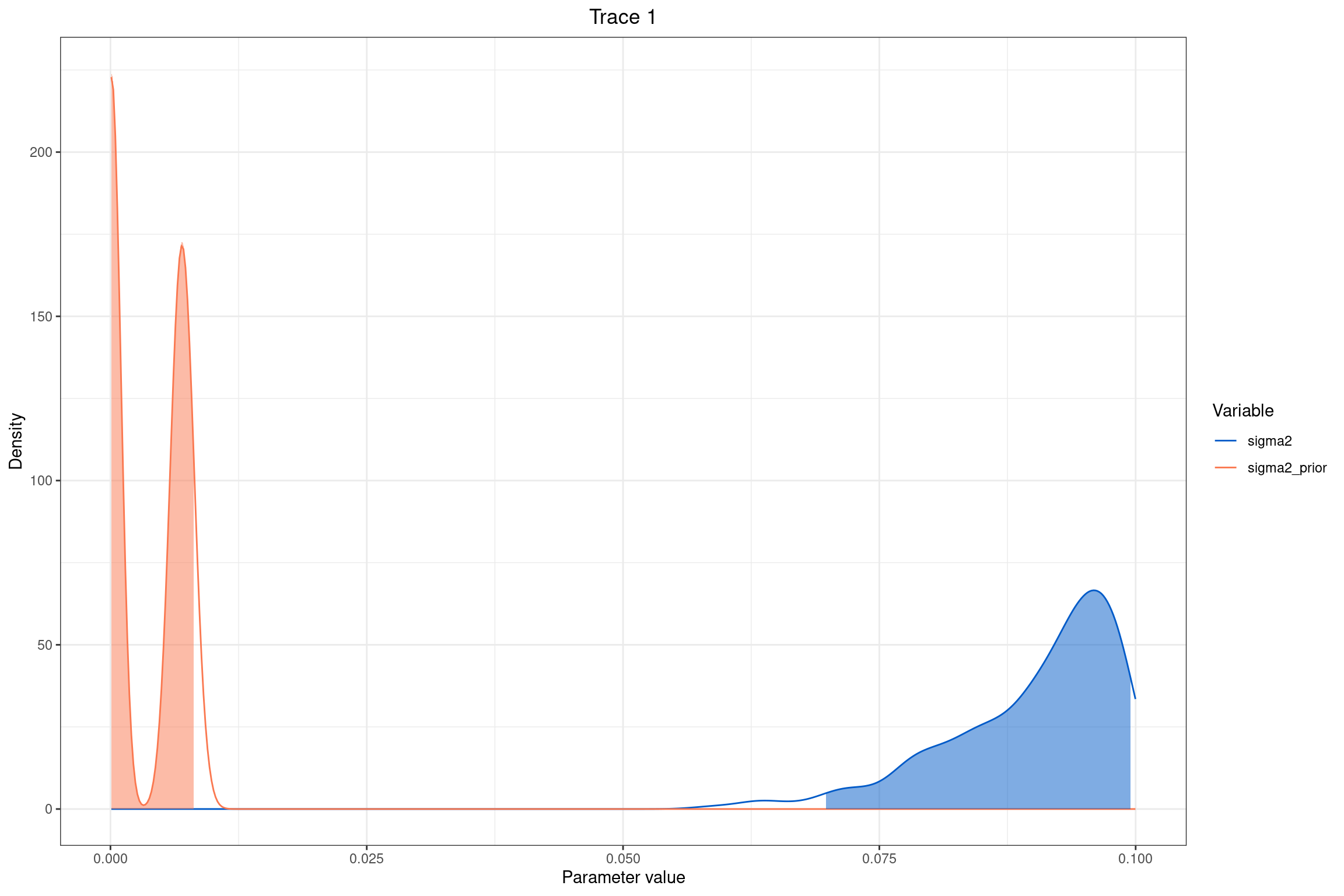
📈 Interpretación de la gráfica de \(\sigma^2\)
library(RevGadgets)library(ggplot2)# Leer archivossimple_BM_posterior <-readTrace("../docs/u1_PatDiv/output/morpho/simple_BM.log")[[1]]simple_BM_prior <-readTrace("../docs/u1_PatDiv/output/morpho/simple_BM_prior.log")[[1]]# Añadir la prior a la tabla posteriorsimple_BM_posterior$sigma2_prior <- simple_BM_prior$sigma2# Graficarplot <-plotTrace(list(simple_BM_posterior), vars=c("sigma2", "sigma2_prior"))plot
[[1]]
Eje X: valores del parámetro \(\sigma^2\) (tasa evolutiva).
Eje Y: densidad de probabilidad.
Línea roja (sigma2_prior): lo que asumías posible antes de ver los datos.
Línea azul (sigma2): lo que los datos te dicen que es más probable.
✅ Interpretación esperada: Si la curva azul (posterior) está desplazada respecto a la roja, significa que tus datos aportaron información nueva.
Si la curva azul es más estrecha, implica que la incertidumbre sobre \(\sigma^2\) se redujo gracias a los datos.
Si las dos curvas son similares, puede que tus datos no sean muy informativos para este modelo.
Modelo Ornstein-Uhlenbeck (OU) simple en RevBayes
El modelo de Ornstein-Uhlenbeck (OU) extiende el modelo de Movimiento Browniano (BM) al asumir que el carácter evoluciona hacia un valor óptimo (θ), en lugar de cambiar al azar.
θ (theta): valor óptimo hacia el cual evoluciona el carácter.
σ²: tasa de variación aleatoria (igual que en BM).
α (alpha): fuerza de atracción hacia el valor óptimo. Cuanto mayor es α, más fuertemente el carácter es “jalado” hacia θ.
A veces se le llama el parámetro de “banda elástica”.
Si α = 0, el modelo OU se reduce al modelo BM.
🔄 Comparación entre script posterior y prior del modelo OU
1️⃣ Script de posterior (mcmc_OU.Rev)
Utiliza tanto el modelo como los datos observados.
Fija los datos con:
X.clamp(data)
🔍 ¿Qué nos dice?
¿Qué valores de los parámetros (alpha, sigma², theta) son más probables después de ver los datos?
Nos permite inferir la historia evolutiva del rasgo bajo un modelo que considera evidencia empírica.
################################################################################# Modelo de Ornstein-Uhlenbeck simple para caracteres continuos# Estimación del valor óptimo evolutivo (theta), la tasa de adaptación (alpha)# y la tasa de evolución estocástica (sigma2) en un árbol calibrado############################################################################################################### Lectura del árbol y datos ###############################trait <-1# Leer el árbol calibradoT <-readTrees("../data/subarbol_ingroup_morph.nex")[1]# Leer la matriz de caracteres continuosdata <-readContinuousCharacterData("../data/med_geiger.nex")data.excludeAll()data.includeCharacter(trait)# Inicializar vectores de movimientos y monitoresmoves =VectorMoves()monitors =VectorMonitors()############################### Modelo del árbol conocido ###############################tree <- T######################### Parámetro sigma² ########################## La tasa estocástica de evolución está controlada por sigma².# Usamos una distribución log-uniforme para representar ignorancia# sobre el orden de magnitud del parámetro.sigma2 ~dnLoguniform(1e-3, 1)moves.append( mvScale(sigma2, weight=2.0) )######################### Parámetro de adaptación alpha ########################## Alpha controla la velocidad de adaptación hacia el valor óptimo.# Usamos una distribución exponencial con media igual a la mitad# de la edad de la raíz dividida por ln(2), lo que implica que# esperamos una "media vida filogenética" igual a la mitad de la edad del árbol.root_age :=tree.rootAge()alpha ~dnExponential( abs(root_age /2.0/ln(2.0)) )moves.append( mvScale(alpha, weight=2.0) )######################### Parámetro óptimo theta ########################## El valor óptimo evolutivo del carácter.# Usamos una distribución uniforme vaga entre -10 y 10.# Dado que puede ser negativo o positivo, usamos un movimiento slide.theta ~dnUniform(-10, 10)moves.append( mvSlide(theta, weight=2.0) )########################################## MCMC más eficiente con movimiento multivariado########################################## Los parámetros del modelo OU pueden estar correlacionados.# Por ello, agregamos un movimiento que aprende la estructura de covarianza# entre sigma2, alpha y theta.avmvn_move =mvAVMVN(weight=5, waitBeforeLearning=500, waitBeforeUsing=1000)avmvn_move.addVariable(sigma2)avmvn_move.addVariable(alpha)avmvn_move.addVariable(theta)moves.append(avmvn_move)################################################### Variables derivadas para interpretar el modelo #################################################### Media vida filogenética: tiempo que tarda un carácter en recorrer# la mitad de la distancia entre el estado de raíz y el óptimot_half :=ln(2.0) / alpha# Reducción de varianza esperada debida a la selección:# compara la varianza bajo selección con la varianza bajo deriva pura (BM)p_th :=1- (1-exp(-2.0* alpha * root_age)) / (2.0* alpha * root_age)############################## Modelo OU con algoritmo REML ############################### Definimos el modelo estocástico para los datos observados bajo OU# Suponemos que el carácter inicia en el óptimo theta en la raízX ~dnPhyloOrnsteinUhlenbeckREML(tree, alpha, theta, sqrt(sigma2), rootStates=theta)# Fijar los datos al modeloX.clamp(data)######################## Crear objeto del modelo ######################### Usamos `model(theta)` aunque también podrías usar `model(sigma2)`mymodel =model(theta)########################### Configurar monitores ############################ Guardar salida del modelo en archivomonitors.append( mnModel(filename="../out/morpho/simple_OU.log", printgen=10) )# Mostrar en pantalla cada 1000 generacionesmonitors.append( mnScreen(printgen=1000, sigma2, alpha, theta) )####################################### Inicializar y ejecutar el MCMC ######################################## Inicializar el MCMC con los movimientos y monitores definidosmymcmc =mcmc(mymodel, monitors, moves, nruns=2, combine="mixed")# Ejecutar el MCMCmymcmc.run(generations=50000)# Salir del entorno de RevBayesq()
2️⃣ Script de prior (mcmc_OU_prior.Rev)
No usa los datos observados.
Omite los datos con:
mymodel.ignoreAllData()
🔍 ¿Qué nos dice?
¿Qué creíamos posible sobre los parámetros (alpha, sigma², theta) antes de ver los datos, solo con la estructura del modelo?
Nos permite explorar los supuestos previos y compararlos con la distribución posterior (para ver cuánto aportaron los datos).
################################################################################# Muestreo desde la distribución a priori del modelo de Ornstein-Uhlenbeck# No se usa la información observada (sin clamp), útil para comparar con posterior############################################################################################################### Lectura del árbol solamente ################################ Leer el árbol filogenético calibradoT <-readTrees("../data/subarbol_ingroup_morph.nex")[1]tree <- T# Inicializar vectores de movimientos y monitoresmoves =VectorMoves()monitors =VectorMonitors()######################### Parámetro sigma² ########################## Tasa estocástica de evoluciónsigma2 ~dnLoguniform(1e-3, 1)moves.append( mvScale(sigma2, weight=2.0) )######################### Parámetro de adaptación alpha ########################## Alpha controla la fuerza de atracción hacia el óptimoroot_age :=tree.rootAge()alpha ~dnExponential( abs(root_age /2.0/ln(2.0)) )moves.append( mvScale(alpha, weight=2.0) )######################### Parámetro óptimo theta ########################## Valor óptimo hacia el que evoluciona el rasgotheta ~dnUniform(-10, 10)moves.append( mvSlide(theta, weight=2.0) )########################################## Movimiento conjunto adaptativo para eficiencia ##########################################avmvn_move =mvAVMVN(weight=5, waitBeforeLearning=500, waitBeforeUsing=1000)avmvn_move.addVariable(sigma2)avmvn_move.addVariable(alpha)avmvn_move.addVariable(theta)moves.append(avmvn_move)################################################### Variables derivadas para interpretación posterior #################################################### Media vida filogenéticat_half :=ln(2.0) / alpha# Reducción porcentual de la varianza debido a selecciónp_th :=1- (1-exp(-2.0* alpha * root_age)) / (2.0* alpha * root_age)############################## Modelo OU con algoritmo REML ############################### Modelo OU completo, pero sin usar datos observadosX ~dnPhyloOrnsteinUhlenbeckREML(tree, alpha, theta, sqrt(sigma2), rootStates=theta)# No usamos X.clamp(data)######################## Crear objeto del modelo ########################mymodel =model(theta)mymodel.ignoreAllData()########################### Configurar monitores ###########################monitors.append( mnModel(filename="../out/morpho/simple_OU_prior.log", printgen=10) )monitors.append( mnScreen(printgen=1000, sigma2, alpha, theta) )####################################### Inicializar y ejecutar el MCMC #######################################mymcmc =mcmc(mymodel, monitors, moves, nruns=2, combine="mixed")mymcmc.run(generations=50000)# Finalizarq()
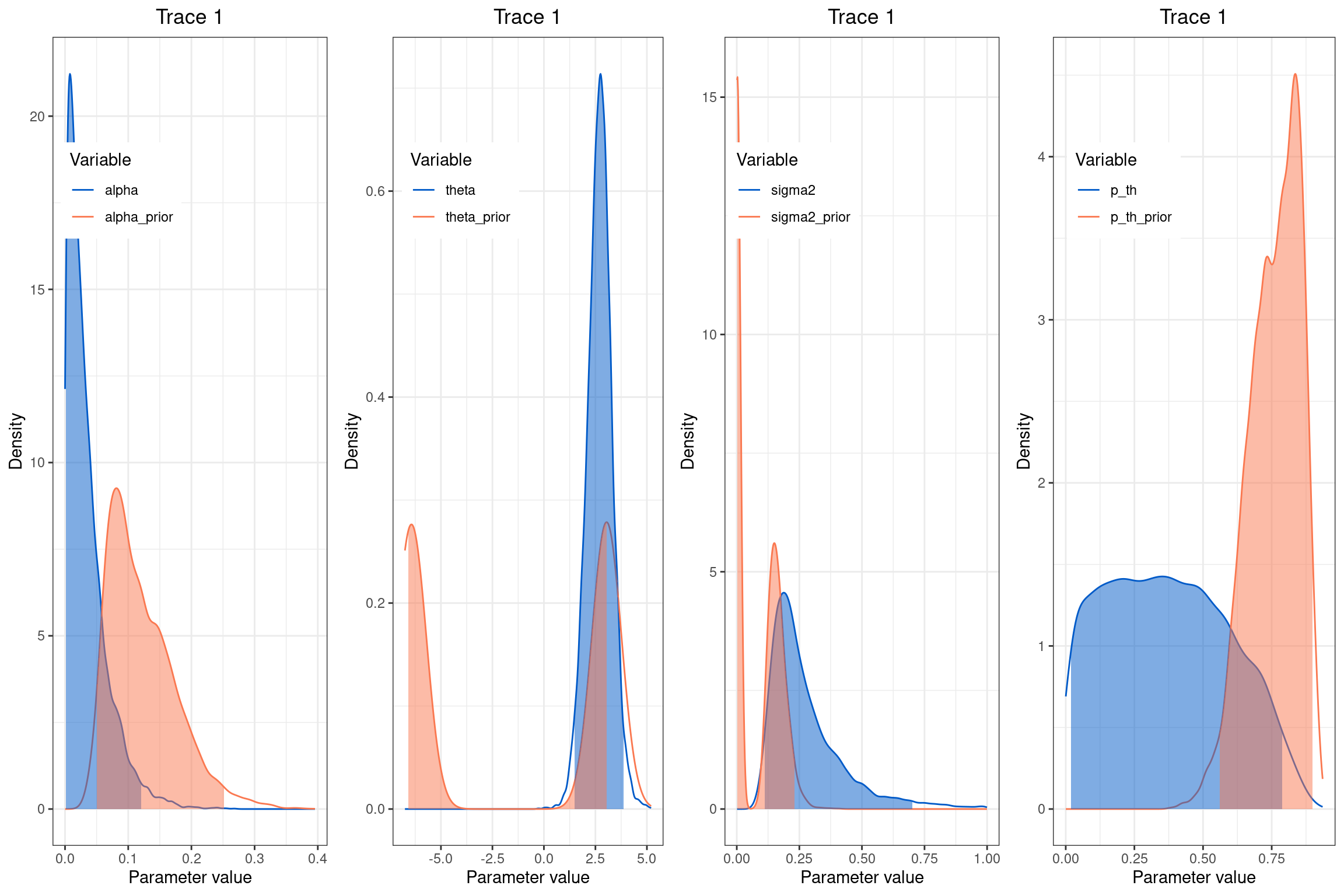
📊 Visualización de parámetros del modelo Ornstein-Uhlenbeck (OU)
# Cargar las librerías para trazar y organizar gráficoslibrary(RevGadgets)library(gridExtra)library(ggplot2)# Leer las muestras de la cadena posterior (después de ver los datos)samples_OU <-readTrace("../docs/u1_PatDiv/output/morpho/simple_OU.log", burnin =0.25)[[1]]# Leer las muestras de la cadena previa (antes de ver los datos)samples_OU_prior <-readTrace("../docs/u1_PatDiv/output/morpho/simple_OU_prior.log", burnin =0.25)[[1]]# Combinar prior y posterior en un solo data framesamples_OU$alpha_prior <- samples_OU_prior$alphasamples_OU$theta_prior <- samples_OU_prior$thetasamples_OU$sigma2_prior <- samples_OU_prior$sigma2samples_OU$t_half_prior <- samples_OU_prior$t_halfsamples_OU$p_th_prior <- samples_OU_prior$p_th# Trazar alpha (fuerza de atracción hacia el óptimo)alpha_plot <-plotTrace(list(samples_OU), vars =c("alpha", "alpha_prior"))[[1]] +theme(legend.position =c(0.25, 0.81))# Trazar theta (valor óptimo del rasgo)theta_plot <-plotTrace(list(samples_OU), vars =c("theta", "theta_prior"))[[1]] +theme(legend.position =c(0.25, 0.81))# Trazar sigma² (tasa estocástica de evolución)sigma_plot <-plotTrace(list(samples_OU), vars =c("sigma2", "sigma2_prior"))[[1]] +theme(legend.position =c(0.25, 0.81))# Trazar t_half (media vida filogenética), usando escala logarítmicat_half_plot <-plotTrace(list(samples_OU), vars =c("t_half", "t_half_prior"))[[1]] +scale_x_log10() +theme(legend.position =c(0.25, 0.81))# Trazar p_th (porcentaje de reducción de varianza por selección)p_th_plot <-plotTrace(list(samples_OU), vars =c("p_th", "p_th_prior"))[[1]] +theme(legend.position =c(0.25, 0.81))# Mostrar una cuadrícula con los parámetros clavegrid.arrange(alpha_plot, theta_plot, sigma_plot, p_th_plot, nrow =1)
📌 ¿Cómo interpretar los gráficos? Cada gráfico muestra dos curvas:
🔵 Posterior (azul): qué valores son más probables después de ver los datos.
🔴 Prior (rojo): qué valores eran posibles antes de ver los datos.
Diferencias claras entre ambas curvas indican que los datos aportaron información real sobre ese parámetro.
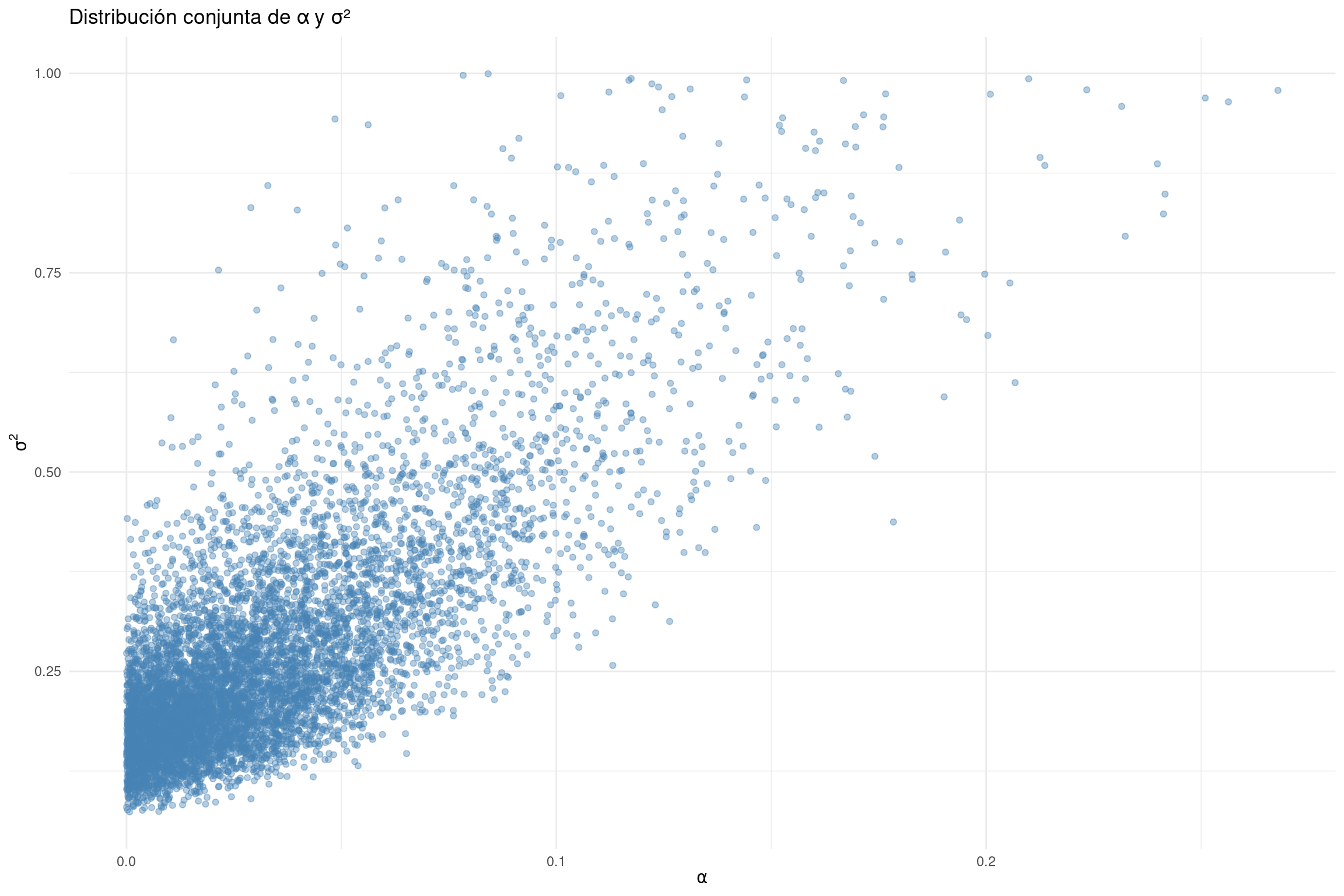
📈 ¿Por qué visualizar la distribución conjunta de alpha y sigma2? En el modelo de Ornstein-Uhlenbeck (OU), los caracteres tienden a una distribución estacionaria que es normal con:
Media: θ (el valor óptimo)
Varianza: \(\sigma^2/(2\alpha)\)
Esto significa que \(\sigma^2\) (la tasa de evolución estocástica) y \(\alpha\) (la fuerza de atracción hacia el óptimo) actúan conjuntamente para determinar la dispersión del rasgo en el tiempo.
⚠️ ¿Cuál es el problema? Si las ramas del árbol son largas o si los datos son poco informativos, es difícil estimar \(\sigma^2\) y \(\alpha\) por separado, porque pueden compensarse entre sí:
Un valor alto de \(\sigma^2\) puede “verse igual” que un valor bajo de \(\alpha\), y viceversa.
Esto se refleja como una correlación negativa entre ambos parámetros en la distribución posterior.
📊 ¿Qué hacer? Graficar la distribución conjunta te permite ver si están correlacionados, lo cual es una señal de que no deberías interpretar σ² o α individualmente.
✅ Código en R para graficar la correlación
library(ggplot2)# Graficar los valores posteriores de alpha vs sigma²ggplot(samples_OU, aes(x=alpha, y=sigma2)) +geom_point(alpha =0.4, color ="steelblue") +theme_minimal() +labs(x =expression(alpha), y =expression(sigma^2),title ="Distribución conjunta de α y σ²")
🧠 Interpretación:
Si los puntos forman una nube diagonal descendente, entonces hay correlación negativa.
Si los puntos forman una nube diagonal ascendente, entonces hay correlación positiva.
En ese caso, no debes interpretar \(\alpha\) o \(\sigma^2\) por separado, porque los datos no permiten distinguir su efecto individual.
Esto es esperado en árboles con ramas largas o con pocos datos informativos.
Modelos de evolución de caracteres discretos
Los modelos de evolución morfológica discreta no solo son útiles para inferir árboles filogenéticos, sino también para formular y poner a prueba hipótesis evolutivas específicas sobre los caracteres de interés. Por ejemplo, podemos comparar distintos modelos de evolución, como modelos reversibles o irreversibles, y estimar las tasas de cambio entre estados. Esto permite evaluar hipótesis como la ley de Dollo, que propone que un carácter complejo puede perderse, pero no recuperarse evolutivamente.
Además, estos modelos nos permiten realizar estimaciones de estados ancestrales, es decir, inferir los posibles estados que tenían los caracteres en los ancestros comunes a partir de los estados observados en las especies actuales. Esta tarea implica integrar (marginalizar) sobre todas las posibles historias evolutivas no observadas que podrían haber ocurrido a lo largo del árbol, dadas las tasas de cambio y la estructura filogenética.
Este enfoque es especialmente poderoso en el marco bayesiano, ya que permite muestrear distribuciones completas de los estados ancestrales y realizar inferencias sólidas sobre la evolución de caracteres morfológicos discretos a lo largo del tiempo.
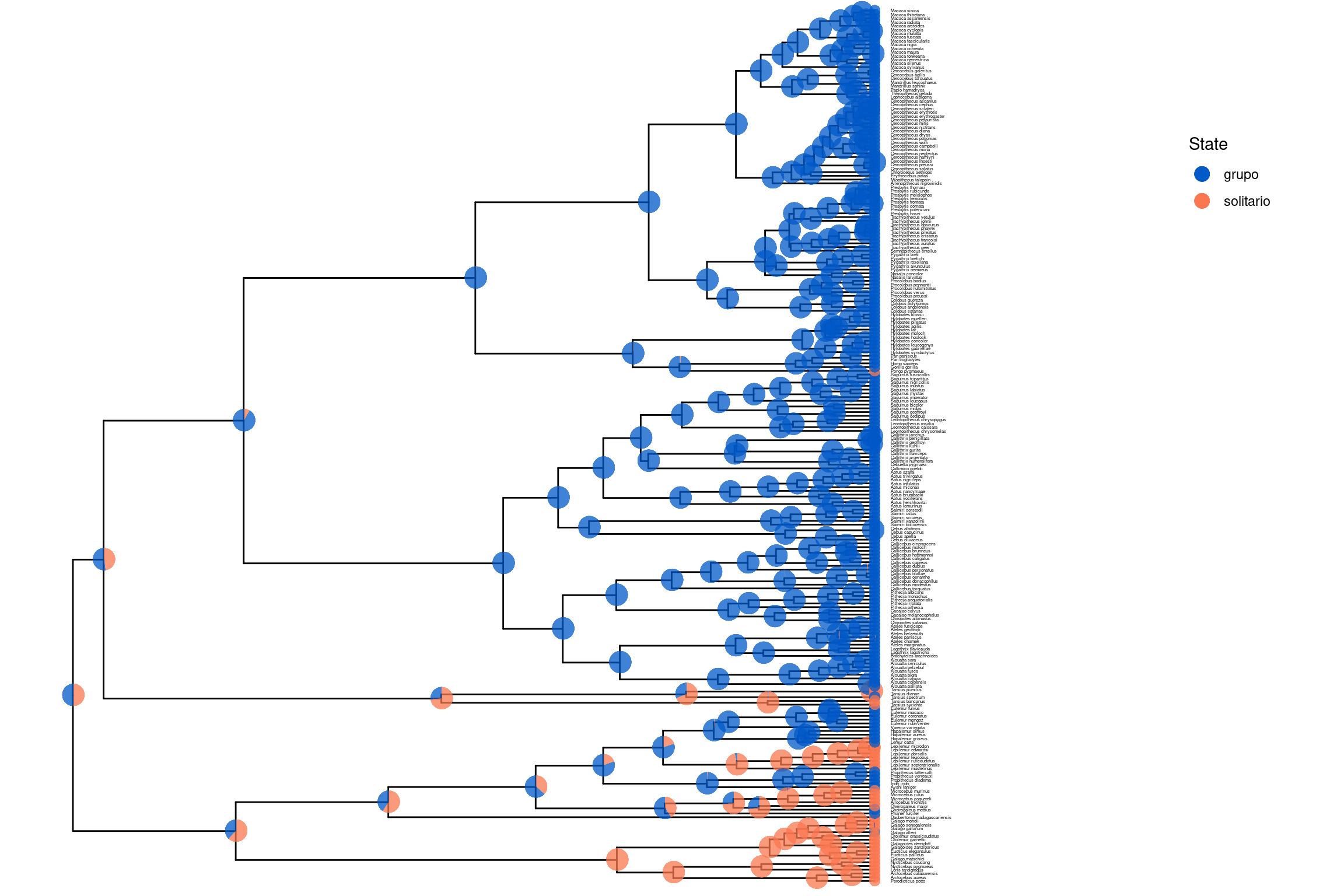
📘 Estimación de estados ancestrales en caracteres morfológicos discretos
En este ejemplo trabajaremos con la evolución de un carácter morfológico discreto en primates: el grado de vida solitaria, con dos posibles estados:
0 = vida en grupo
1 = vida solitaria.
Nuestro objetivo principal es inferir el estado ancestral de este carácter en el ancestro común de todos los primates. Además, queremos evaluar si la evolución de este rasgo sigue un modelo con tasas de transición iguales (ERM), tasas desiguales, o incluso un modelo irreversible (por ejemplo, si una vez que se adopta la vida solitaria, no se vuelve a la vida grupal).
Para comenzar, especificaremos un modelo ERM de dos estados (k = 2), también conocido como modelo de tasas iguales de Markov (Pagel 1994; Lewis 2001). En este modelo, la tasa de cambio entre cualquier par de estados es la misma. La matriz de tasas \(Q\) se simplifica a:
Este modelo es análogo al modelo de Jukes-Cantor para datos moleculares, donde no se estima la frecuencia de cada estado, sino que se asume que todos los estados tienen la misma probabilidad a priori.
Este enfoque nos permite explorar, de manera bayesiana, cómo han evolucionado los caracteres discretos y cuáles fueron los estados más probables en los ancestros, dado un modelo evolutivo simple y los datos observados en las especies actuales.
Descarga del Árbol Filogenético y la matriz de caracteres discretos
📥 Puedes descargar el archivo .tre con el árbol en el siguiente enlace:
Guarda los archivos en la carpeta correspondiente y verifica su ubicación antes de continuar.
################################################################################}# Ejemplo en RevBayes: Inferencia de estados ancestrales y tasas de evolución # morfológica bajo un modelo de tasas iguales de Markov (ERM)############################################################################################################## Lectura de los datos ############################### Nombre del carácter morfológico a analizar (modificable)CHARACTER ="solitariness"NUM_STATES =2# Importar la matriz de datos morfológicos discretosmorpho <-readDiscreteCharacterData("data/primates_"+ CHARACTER +".nex")# Crear vectores para movimientos y monitores del MCMCmoves =VectorMoves()monitors =VectorMonitors()#################### Leer el árbol ##################### Leer el árbol filogenético (en este caso, uno fijo)# Nota: readTrees devuelve un vector de árboles, por eso tomamos el primerophylogeny <-readTrees("data/primates_tree.nex")[1]################################### Especificar el modelo de tasas #################################### Definir una tasa global μ con distribución exponencial# La media está basada en la longitud del árbol (10 eventos esperados)rate_pr :=phylogeny.treeLength() /10mu ~dnExp(rate_pr)# Movimiento para proponer nuevos valores de mu durante el MCMCmoves.append( mvScale(mu, weight=2) )# Crear vector de tasas (en el modelo ERM todas las tasas son iguales)NUM_RATES = NUM_STATES * (NUM_STATES -1)for (i in1:NUM_RATES) { rate[i] := mu}################################### Definir la matriz de tasas Q #################################### Generar la matriz de tasas para el modelo de caracteres morfológicosQ_morpho :=fnFreeK(rate, rescale=false)########################################### Especificar frecuencias en la raíz ############################################ Usamos una distribución uniforme (1,1) como prior para los estados en la raízrf_prior <-rep(1, NUM_STATES)rf <-simplex(rf_prior)######################### Modelo evolutivo CTMC ########################## Definir el modelo de evolución morfológica bajo un proceso CTMCphyMorpho ~dnPhyloCTMC(tree=phylogeny, Q=Q_morpho, rootFrequencies=rf, type="Standard")# Fijar los datos observados al modelophyMorpho.clamp(morpho)########################## Crear objeto del modelo ########################### Crear el objeto global del modelo a partir del árbolmymodel =model(phylogeny)########################## Especificar monitores ########################### 1. Monitor del modelo completo (mu, etc.)monitors.append( mnModel(filename="output/"+ CHARACTER +"_ERM.log", printgen=1) )# 2. Monitor en pantalla para seguimiento en vivomonitors.append( mnScreen(printgen=100) )# 3. Monitor de estados ancestrales condicionales conjuntosmonitors.append( mnJointConditionalAncestralState(tree=phylogeny,ctmc=phyMorpho,filename="output/"+ CHARACTER +"_ERM.states.txt",type="Standard",printgen=1,withTips=true,withStartStates=false) )################################ Configurar y correr el MCMC ################################# Inicializar el objeto MCMCmymcmc =mcmc(mymodel, monitors, moves, nruns=2, combine="mixed")# Ejecutar la cadena MCMC por 25,000 generacionesmymcmc.run(generations=25000, tuningInterval=200)######################################### Procesar y guardar estados ancestrales ########################################## Leer el archivo de estados ancestrales estimadosanc_states =readAncestralStateTrace("output/"+ CHARACTER +"_ERM.states.txt")# Crear árbol anotado con probabilidades posteriores de estados ancestralesanc_tree =ancestralStateTree(tree=phylogeny,ancestral_state_trace_vector=anc_states,include_start_states=false,file="output/"+ CHARACTER +"_ase_ERM.tree",burnin=0.25,summary_statistic="MAP",site=1)# Salir de RevBayesq()
🧬 Visualización de estados ancestrales en R con RevGadgets
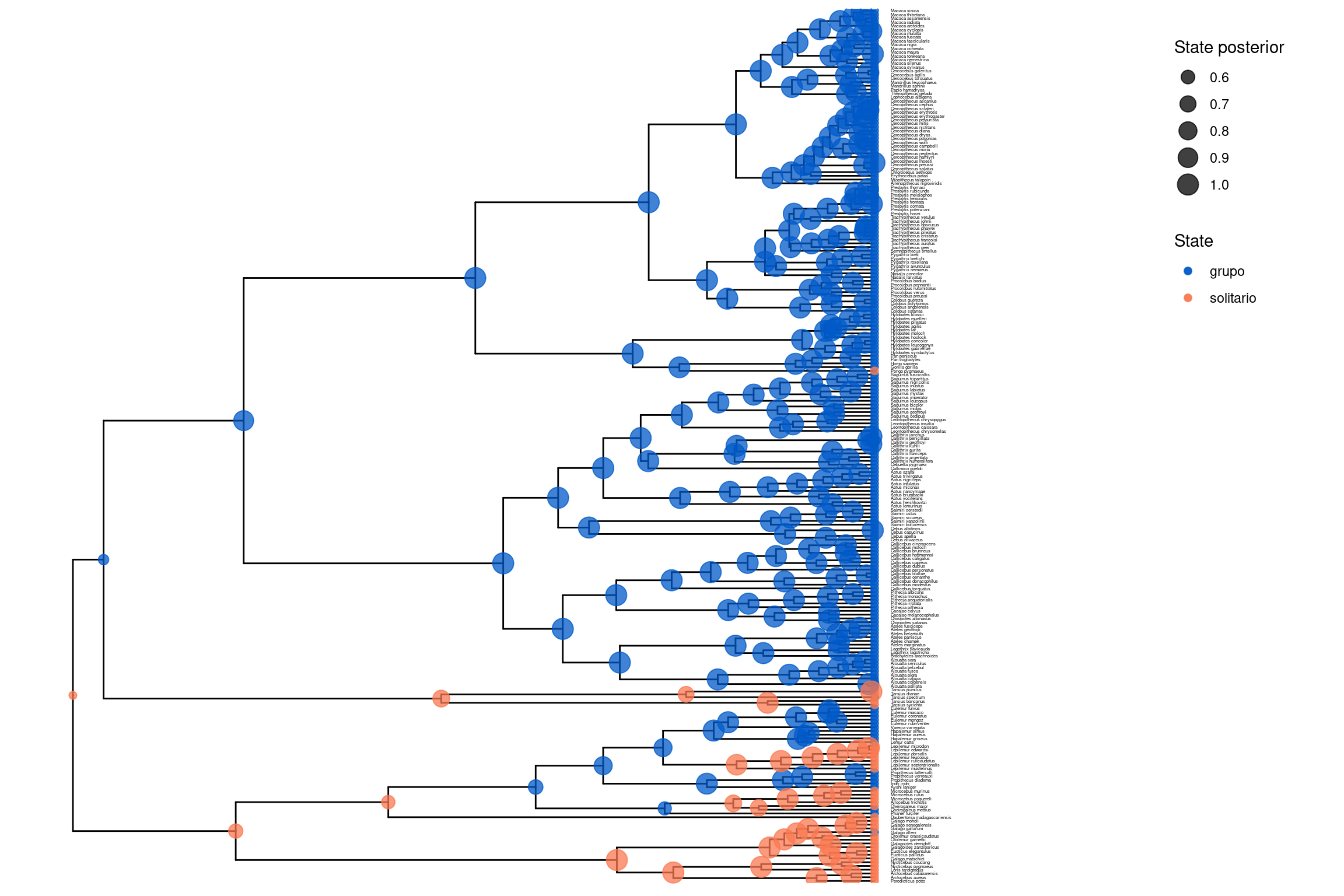
# Cargar la librería necesarialibrary(RevGadgets)library(ggplot2)# Especificar el archivo del árbol con estados ancestralestree_file <-"../docs/u1_PatDiv/output/morpho/solitariness_ase_ERM.tree"# Especificamos los nombres de los estados que se mostrarán en el árbol. Puedes modificarlos según los caracteres analizados.ase <-processAncStates(tree_file,state_labels =c("0"="grupo", "1"="solitario"))
# Graficar los estados ancestrales como colores en nodos (MAP)# Cada nodo del árbol mostrará su estado más probable (MAP).# El color representa el estado, y el tamaño del nodo representa su probabilidad posteriorp1 <-plotAncStatesMAP(t = ase,tree_layout ="rect",tip_labels_size =1) +theme(legend.position =c(0.92, 0.81))p1
# Guardar la figura en PDFggsave("primates_solitariness_ASE_ERM_MAP.pdf", p1, width =11, height =9)# (Opcional) Mostrar incertidumbre con gráficos de pastelp2 <-plotAncStatesPie(t = ase,tree_layout ="rect",tip_labels_size =1) +theme(legend.position =c(0.92, 0.81))p2
# Guardar la figura en PDFggsave("primates_solitariness_ASE_ERM_MAP_2.pdf", p2, width =11, height =9)