Aquí pasamos de solo ver el árbol a interactuar con él.
La manipulación nos permite explorar mejor la topología y preparar figuras más limpias:
👁️ Visualizar árbol con ggtree
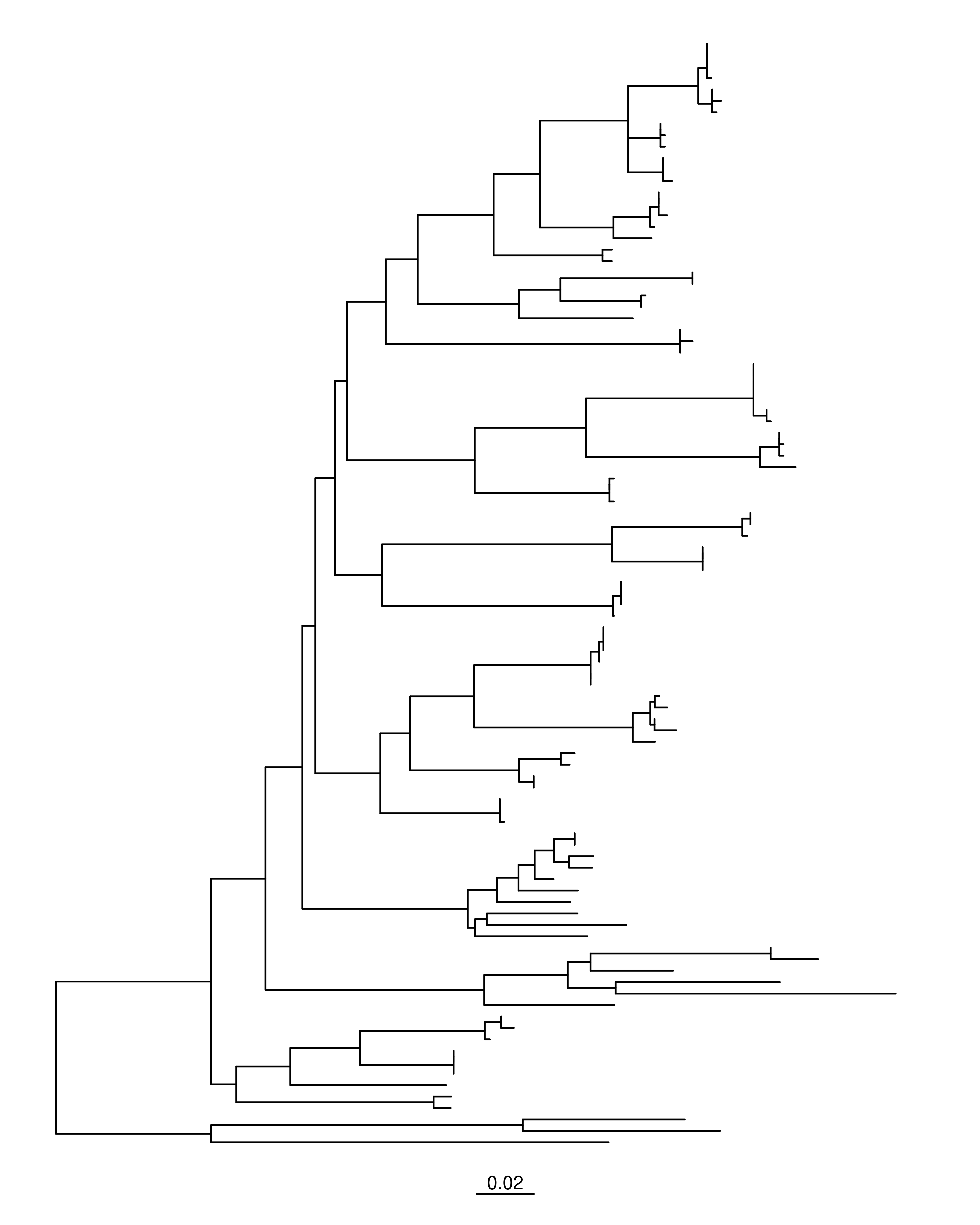
## Paqueteslibrary(tidyverse)library(ggtree)library(treeio)library(ape)# Archivo ML (NEWICK)f_raxml <-"../docs/raxml_cox1_Stenopelmatus.tre"# Lectura con apeml_phylo <-read.tree(f_raxml)# geom_tiplab() agregra nombres de terminales# size controla el tamaño del textoml_p <-ggtree(ml_phylo) ml_p
📊 El “data” de un objeto phylo
Cuando cargamos un árbol en R con ape lo que tenemos es un objeto de clase phylo.
Este objeto contiene varias piezas internas:
# Estructura básica de un árbol phylostr(ml_phylo)
🏷️ label → Nombre del taxón (si es tip) o valor de soporte (si es nodo).
✅/❌ isTip → TRUE si es terminal, FALSE si es nodo interno.
📐 x, y → Coordenadas asignadas por ggtree para graficar.
🔀 branch → Longitud usada para posicionar gráficamente.
📐 angle → Ángulo de la rama (en layouts circulares o fan).
🔗 Integrar metadatos al árbol con el operador %<+% de ggtree
💾 Metadatos para el árbol (qué y para qué)
A veces necesitamos información extra para cada terminal (muestra) que no viene en el árbol:
Asignación taxonómica: especie, género, clado, área biogeográfica, etc.
Rasgos morfológicos: presencia/ausencia (0/1), conteos (número de espinas), medidas (mm), etc.
Datos ecológicos: hábitat, elevación, bioma, etc.
📌 Integrar metadatos al árbol con %<+%
Los metadatos se integran al gráfico con el operador %<+%, que funciona como un “enchufe”:
Fusiona por la columna label.
Esa columna debe estar en tu dataframe de metadatos y debe coincidir exactamente con los nombres de las terminales del árbol (ml_p$data$label).
👉 Es decir, el df con tus metadatos necesita una columna llamada label, donde cada valor sea idéntico al nombre de las puntas del árbol.
✅ Cuándo usar %<+%
Cuando tus IDs de tips en el árbol y tu tabla coinciden 1:1 (mismo texto).
Cuando quieres evitar hacer joins manuales con dplyr y trabajar de inmediato con geom_* de ggtree.
🧩 Requisitos mínimos
La tabla debe tener una columna con los mismos nombres que label del árbol.
Si tu columna no se llama label, renómbrala en tu tabla.
# Cargar la tabladf_stenopelmatus <-read.csv("../docs/stenopelmatus.csv") %>%rename(label = Sample) # renombrar las columna Sample por labelhead(df_stenopelmatus)
label Lat Long Species Text
1 CNIN3620 15.649 -92.809 Stenopelmatus_micropterous 40: S. sp. aff. chiapas
2 CNIN3621 15.649 -92.809 Stenopelmatus_micropterous <NA>
3 CNIN3624 15.722 -92.939 Stenopelmatus_macropterous 25: S. sartorianus
4 CNIN3625 15.722 -92.939 Stenopelmatus_macropterous <NA>
5 CNIN3626 15.722 -92.939 Stenopelmatus_macropterous <NA>
6 CNIN3630 19.631 -97.009 Stenopelmatus_apterous <NA>
Color Presencia_alas Region
1 negro no Mexico: Chiapas Highlands province
2 negro no Mexico: Chiapas Highlands province
3 naranja si Mexico: Chiapas Highlands province
4 naranja si Mexico: Chiapas Highlands province
5 naranja si Mexico: Chiapas Highlands province
6 rojo no Mexico: Transmexican Volcanic Belt province
#Unir árbol + informacion extra ml_p <- ml_p %<+% df_stenopelmatus# Transforma en un data.frameml_df <- ml_p$datahead(ml_df[,4:12])
# ggtree(...)$data expone la tabla interna con columnas como x, y, node, isTip, labelggtree(ml_phylo) +geom_text2(aes(label = node),hjust =-0.1, size =2.5, color ="gray25")
👉 Tip: los números de nodos no aparecen en el Newick, son generados por R al leer el árbol.
Los tips siempre se numeran del 1 al número de taxa.
Los nodos internos siguen con números consecutivos.
🔄Rotar el árbol
¿Por qué rotar un árbol?
No cambia la topología: rotar solo invierte el orden de los dos subclados hijos de un nodo interno. Las relaciones evolutivas permanecen idénticas.
Mejora la legibilidad: puedes alinear clados de interés, reducir cruces de ramas y acomodar etiquetas para una figura más limpia.
Estética para publicación: ayuda a que comparaciones (p. ej., entre clados hermanos) queden “lado a lado” como quieres mostrar en el manuscrito.
Ejemplo
# Rotar en el nodo 116 p_rotate_116 <-ggtree(ml_phylo) + ggtree::rotate(node =116) # ggtree guarda el arbol sin rotar y el rotado en una lista# Árbol sin rotarp_rotate_116[[1]] +geom_text2(aes(label = node),hjust =-0.1, size =2.5, color ="gray25")
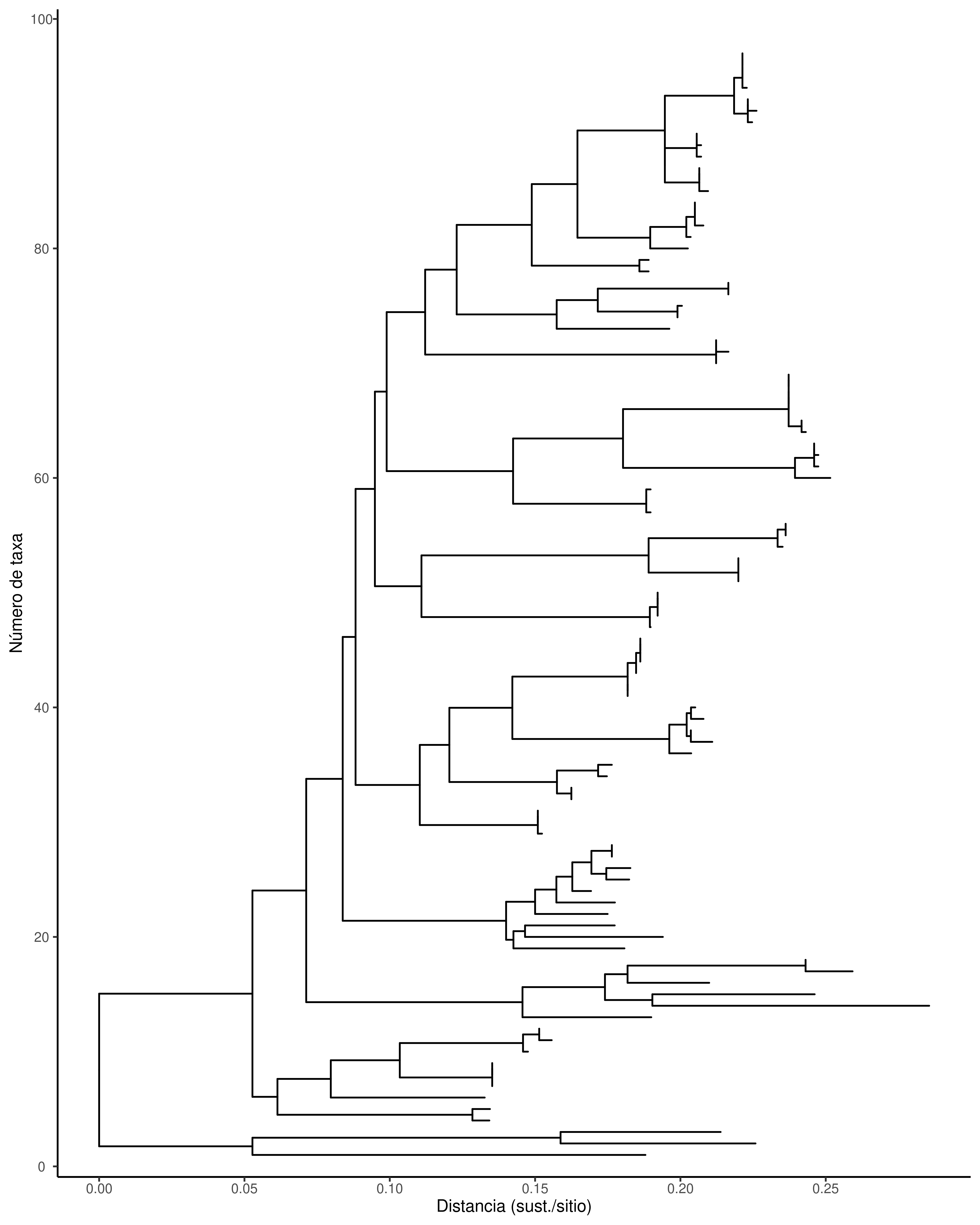
Eje X (x) → representa la distancia acumulada desde la raíz.
Si tu árbol es de Máxima Verosimilitud o inferencia bayesiana, X suele ser sustituciones por sitio.
Si es ultramétrico (ej. BEAST), X puede interpretarse como tiempo.
Eje Y (y) → es un índice que asigna ggtree automáticamente a cada terminal, de arriba a abajo.
No es biológico, solo indica la posición vertical de cada taxón.
# Rango en X (distancia/tiempo)# xr → te dice de dónde a dónde llega el árbol en horizontal.xr <-range(ml_df$x, na.rm =TRUE)# Rango en Y (posición de tips/nodos)# yr → te da el número mínimo y máximo de posiciones verticales (básicamente cuántos tips).yr <-range(ml_df$y, na.rm =TRUE)# 5% de aire a izquierda/derechapad_left <-0.05*diff(xr)pad_right <-0.05*diff(xr)ggtree(ml_phylo) +# Eje X visible y ajustadoscale_x_continuous(name ="Distancia (sust./sitio)", # cambia el nombre si es tiempobreaks =pretty(xr, n =6),limits =c(xr[1] - pad_left, xr[2] + pad_right),expand =c(0, 0) # usamos limits, no expand ) +# Eje Y visible con ticksscale_y_continuous(name ="Número de taxa",breaks =pretty(yr, n =5),expand =expansion(mult =c(0.02, 0.04)) ) +# Mostrar ticks y líneas (ggtree los oculta por default)theme(axis.line.y =element_line(),axis.ticks.y =element_line(),axis.text.y =element_text(size =8),axis.title.y =element_text(size =10),axis.line.x =element_line(),axis.ticks.x =element_line(),axis.text.x =element_text(size =8),axis.title.x =element_text(size =10) )
👉 Para que las etiquetas no se corten, el árbol sea legible y la figura mantenga proporciones al exportar.
🔑 Pasos básicos:
📐 Calcular rangos en x y y desde ggtree(tr)$data.
➕ Agregar un poco de aire a la derecha (pad_x) para las etiquetas.
📏 Usar scale_x_continuous y scale_y_continuous con expand=0 para fijar límites exacto
✂️ coord_cartesian(clip="off") → deja que sobresalgan etiquetas largas.
↔︎️ plot.margin → agrega margen extra si se cortan textos.
📊 geom_treescale → coloca una barra de escala en coordenadas reales.
👉 En pocas líneas: controlas el espacio visible, evitas recortes y aseguras consistencia en tus figuras.
# obtener el numero de terminalesntips_s <- ml_df %>%filter(isTip =="TRUE") %>%nrow()# Aire extra en X (5% del rango) para no cortar etiquetaspad_x <-0.05*diff(xr)ml_p_adj <- ml_p +geom_treescale(y =-3.5, width =0.02) +# escala sustituciones por sitio.scale_x_continuous(limits =c(xr[1] - pad_x, xr[2] + pad_x),expand =c(0, 0)) +# Limites exactos sin padding automáticoscale_y_continuous(limits =c(-5, ntips_s +3),expand =c(0, 0)) +coord_cartesian(clip ="off") +# Permitir etiquetas largas fuera del paneltheme(plot.margin =margin(5.5, 25, 5.5, 5.5)) # Más aire a la derechaml_p_adj
🛡️Soportes de nodos
En un árbol de Máxima Verosimilitud (ML), los nodos internos suelen llevar valores de bootstrap que indican qué tan confiable es cada agrupación:
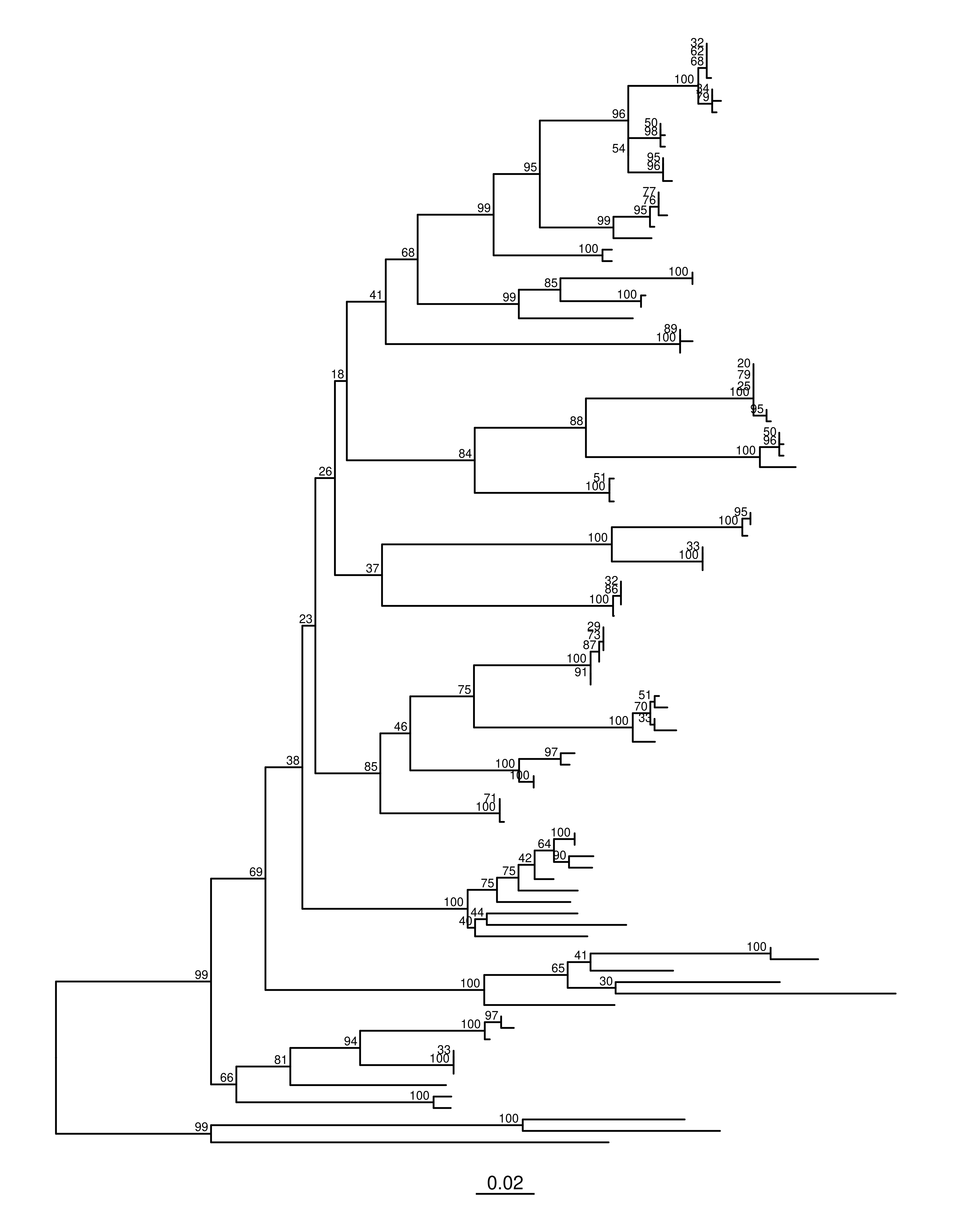
🔢 Primero podemos mostrar los valores en crudo (números en cada nodo).
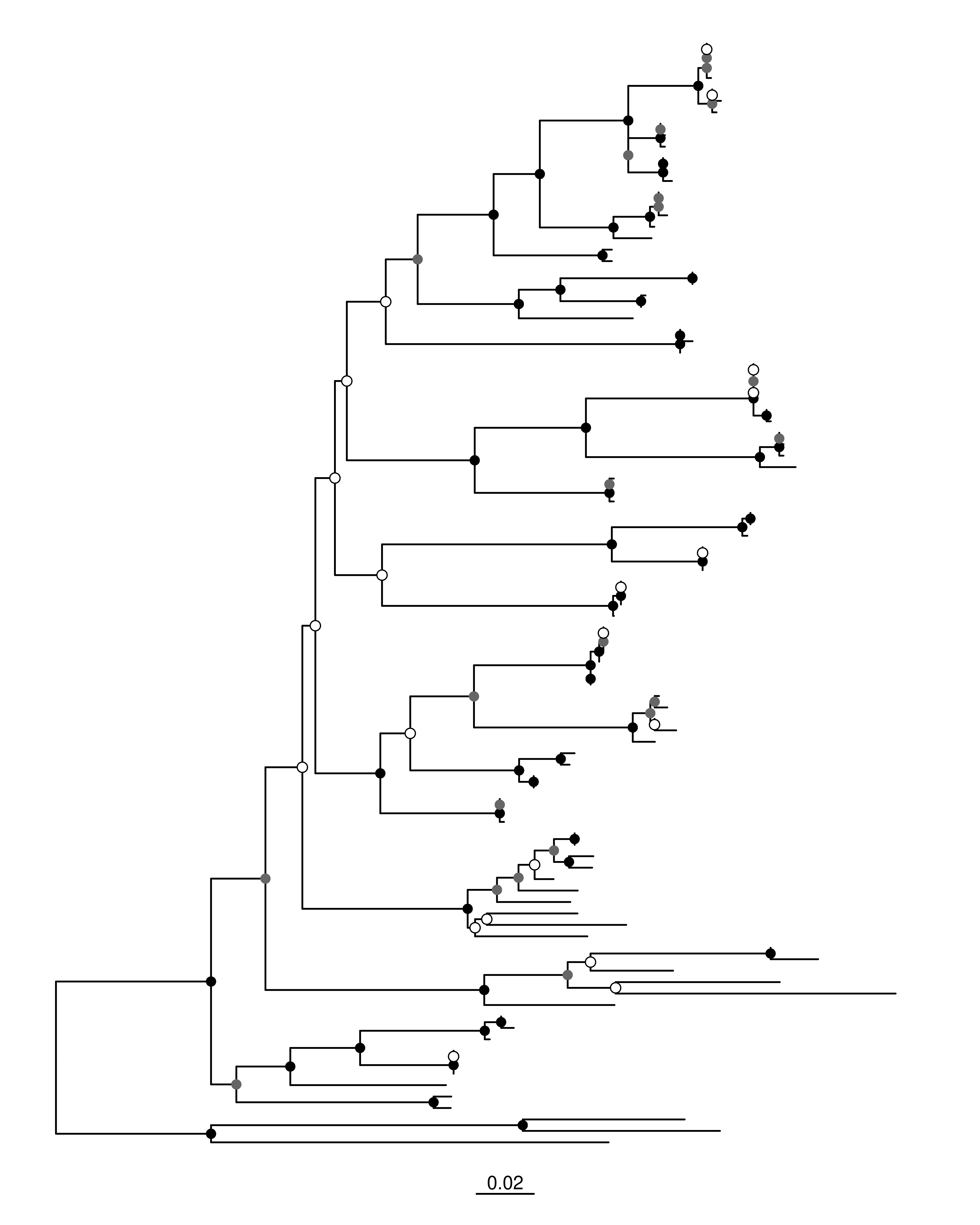
⚫ Después, para figuras más limpias, podemos resumir gráficamente los soportes de acuerdo con Alfaro et al. (2003):
👉 Esto permite pasar de una visualización informativa (con números) a una visualización estética y clara.
A) Mostrar soportes “en crudo”
# Soportes tal cual vienen del árbol (RAxML suele guardarlos en node.label)ml_p_adj +geom_text2(aes(label = label, subset =!isTip & label !=""),size =2.5, vjust =-0.3, hjust =1.2)
B) Puntos según umbrales de bootstrap
> 80: círculos negros
50–80: círculos grises
< 50: círculos blancos
# Puntos por umbral de bootstrapml_p_adj <- ml_p_adj +# Negro: >80geom_point2(aes(subset =!isTip &as.numeric(label) >80),size =2.5, shape =16, color ="black") +# Gris: 50–80geom_point2(aes(subset =!isTip &as.numeric(label) >=50&as.numeric(label) <=80),size =2.5, shape =16, color ="grey40") +# Blanco: <50geom_point2(aes(subset =!isTip &as.numeric(label) <50),size =2.5, shape =21, fill ="white", color ="black")ml_p_adj
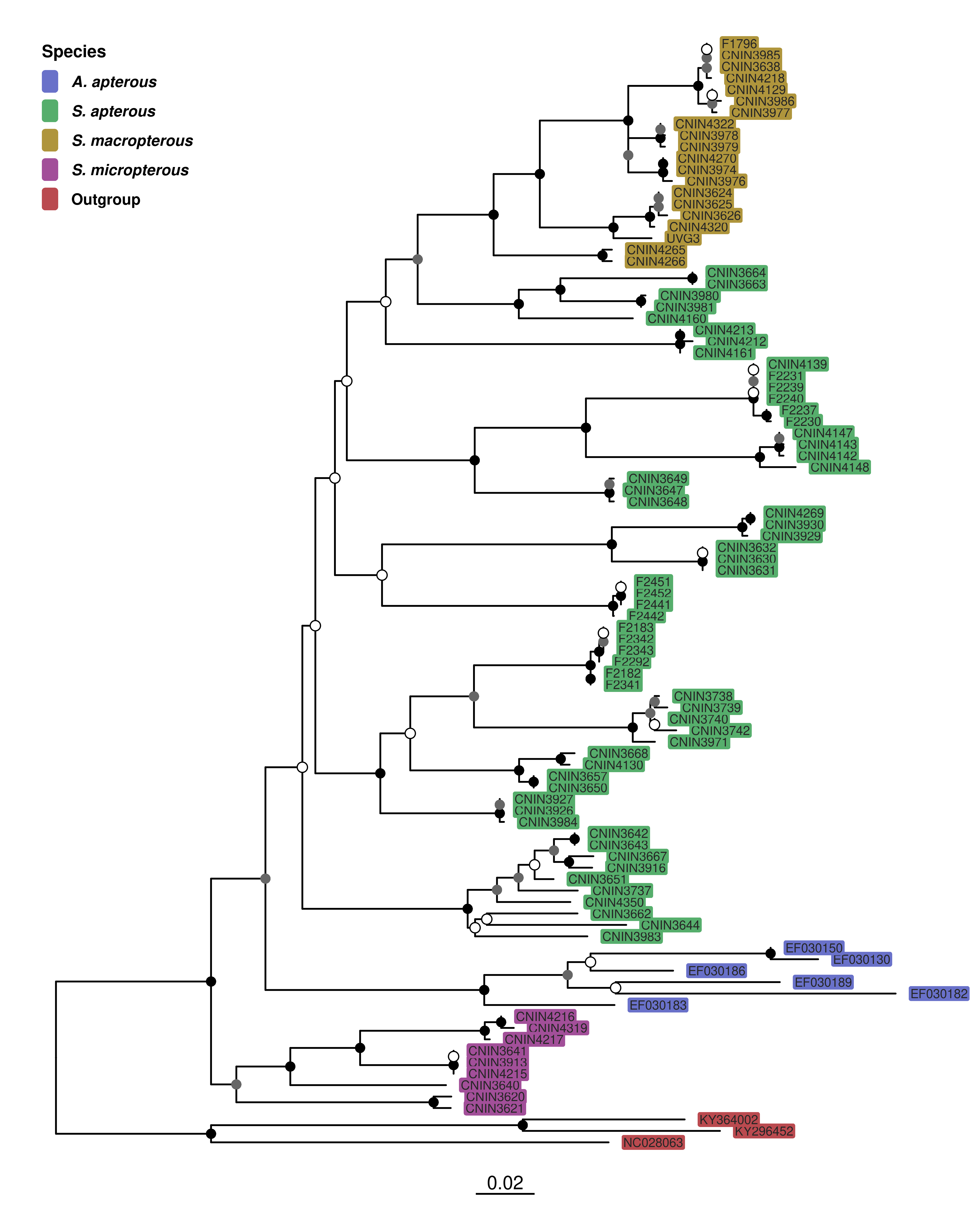
🎨 Etiquetas de las terminales
Hasta ahora solo hemos visto el árbol como estructura y hemos manipulado nodos o tamaños. En esta parte vamos a enriquecerlo con nuestros metadatos —que añadimos en el paso previo con el operador %<+% de ggtree— para poder representar información extra en el árbol.
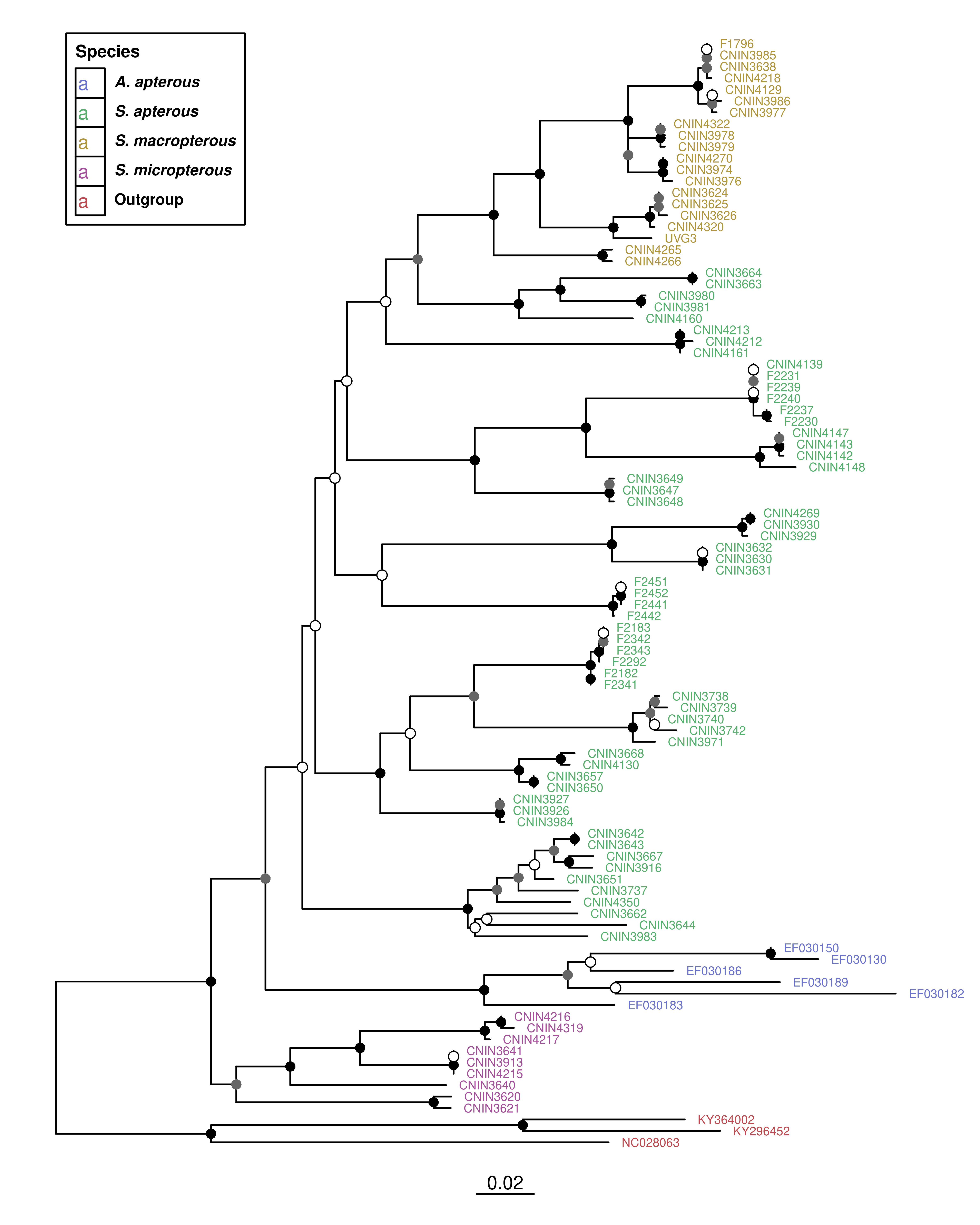
🔤 Pintar los nombres de las terminales por categorías (ej. S. apterous, S. micropterous…).
# Paleta por categoría funcional (ajústala si quieres)pal <-c("Stenopelmatus_apterous"="#56ae6c","Stenopelmatus_micropterous"="#a24f99","Stenopelmatus_macropterous"="#af953c","Ammopelmatus_apterous"="#6971c9","Outgroup"="#ba4a4f")ml_p_adj +geom_tiplab(aes(color = Species), size =2.5, offset =0.003) +scale_color_manual(values = pal,na.value ="black",# orden explícito de los gruposbreaks =c("Ammopelmatus_apterous","Stenopelmatus_apterous", "Stenopelmatus_macropterous","Stenopelmatus_micropterous","Outgroup"),labels =c(Ammopelmatus_apterous =expression(bolditalic("A. apterous")),Stenopelmatus_apterous =expression(bolditalic("S. apterous")),Stenopelmatus_macropterous =expression(bolditalic("S. macropterous")),Stenopelmatus_micropterous =expression(bolditalic("S. micropterous")),Outgroup =expression(bold("Outgroup")))) +guides(color =guide_legend(title ="Species",override.aes =list(size =4))) +theme(legend.position =c(0.25, 0.98), # esquina superior derecha (98% ancho/alto)legend.justification =c("right", "top"), # anclar a esa esquinalegend.background =element_rect(fill ="white", color ="black"), # recuadrolegend.key =element_rect(fill ="white", color ="black"), # caja de cada colorlegend.title =element_text(size =10, face ="bold"),legend.text =element_text(size =9))
🖼️ Resaltar clados completos con recuadros de color (highlight).
# pal: tu paleta por especie (ya definida antes)ml_p_adj +geom_tiplab(aes(fill = Species, label = label), # fill por especiecolor ="grey15",geom ="label", # cajita detrás del textolabel.size =0, # sin borde en la cajitalabel.padding =unit(0.12, "lines"), # relleno interno de la cajitasize =2.6,offset =0.003,show.legend =TRUE ) +scale_fill_manual(values = pal,na.value ="black",# orden explícito de los gruposbreaks =c("Ammopelmatus_apterous","Stenopelmatus_apterous", "Stenopelmatus_macropterous","Stenopelmatus_micropterous","Outgroup"),labels =c(Ammopelmatus_apterous =expression(bolditalic("A. apterous")),Stenopelmatus_apterous =expression(bolditalic("S. apterous")),Stenopelmatus_macropterous =expression(bolditalic("S. macropterous")),Stenopelmatus_micropterous =expression(bolditalic("S. micropterous")),Outgroup =expression(bold("Outgroup")))) +guides(fill =guide_legend(title ="Species",# símbolo cuadrado, sin letra y un poco más pequeño para que el gap sea menoroverride.aes =list(shape =15, size =4, label =NULL, colour =NA, stroke =0),label.hjust =0, title.hjust =0)) +theme(legend.position =c(0.02, 0.98),legend.justification =c("left", "top"),legend.background =element_rect(fill ="white", color ="white"),# menos espacio entre key y textolegend.title =element_text(size =10, face ="bold"),legend.text =element_text(size =9, margin =margin(l =0)) )