En biología evolutiva, no solo nos interesa cómo se relacionan las especies, sino también cuándo divergieron.
Los árboles datados estimados con programas como BEAST incorporan información temporal (fósiles, tasas de sustitución, calibraciones) para asignar edades a los nodos del árbol.
El resultado no es un único valor fijo, sino una distribución de edades y tasas que refleja la incertidumbre inherente al proceso evolutivo.
Por eso, los árboles datados suelen incluir:
📏 Edades de nodos → reportadas como medianas y con intervalos HPD al 95%.
🔄 Longitudes de rama → expresadas generalmente en unidades de tiempo.
⏱️ Tasas de evolución → estimadas sobre todo bajo relojes relajados.
✅ Soportes posteriores → probabilidades a posteriori que indican el grado de confianza en cada clado.
En R, al importar un árbol anotado desde BEAST, toda esta información se organiza en el objeto tree$data, que actúa como una tabla con variables clave para analizar, visualizar y anotar el árbol.
🧰 Preparación de los datos
Primero cargamos los metadatos de las muestras y renombramos la columna Sample a label para que coincida con las etiquetas de las puntas del árbol (tip labels). Luego leemos el árbol anotado por BEAST.
library(treeio)library(ggtree)library(tidyverse)library(deeptime)# Metadata de las muestrasdf_stenopelmatus <-read.csv("../docs/stenopelmatus.csv") %>%rename(label = Sample) # renombra Sample → label para que haga matchhead(df_stenopelmatus)
label Lat Long Species Text
1 CNIN3620 15.649 -92.809 Stenopelmatus_micropterous 40: S. sp. aff. chiapas
2 CNIN3621 15.649 -92.809 Stenopelmatus_micropterous <NA>
3 CNIN3624 15.722 -92.939 Stenopelmatus_macropterous 25: S. sartorianus
4 CNIN3625 15.722 -92.939 Stenopelmatus_macropterous <NA>
5 CNIN3626 15.722 -92.939 Stenopelmatus_macropterous <NA>
6 CNIN3630 19.631 -97.009 Stenopelmatus_apterous <NA>
Color Presencia_alas Region
1 negro no Mexico: Chiapas Highlands province
2 negro no Mexico: Chiapas Highlands province
3 naranja si Mexico: Chiapas Highlands province
4 naranja si Mexico: Chiapas Highlands province
5 naranja si Mexico: Chiapas Highlands province
6 rojo no Mexico: Transmexican Volcanic Belt province
# Leer el árbol anotado por BEASTbeast_tree <-read.beast("../docs/Beast_Stenopelmatinae_cox1.tre")head(beast_tree@phylo$tip.label)
Normalizamos las etiquetas del árbol de BEAST eliminando sufijos que no forman parte del identificador de la muestra (por ejemplo, el marcador _Cox1 o _Cox1p) y versiones agregadas automáticamente (como .1, .2).
# Quitar sufijos "_cox1" o "_cox1p" de los tipsbeast_tree@phylo$tip.label <- beast_tree@phylo$tip.label %>%str_remove("_Cox1p?$") %>%# quita _Cox1 o _Cox1pstr_remove("\\.\\d+$") # quita punto y dígitos al final (.1, .2, etc.)head(beast_tree@phylo$tip.label)
Esta limpieza garantiza que los nombres en label y en los tip labels del árbol coincidan 1:1, lo cual es imprescindible para fusionar metadatos al árbol (p. ej., con %<+% en ggtree) y evitar empates fallidos o duplicados.
📊 El “data” de un objeto treedata
Para explorar y anotar un árbol datado, primero anclamos el eje temporal al presente (0) desplazando el árbol por la edad de la raíz; así el tiempo queda en [-root_age, 0]. Luego integramos los metadatos de las muestras y extraemos beast_p$data en beast_df.
# 1) Graficar el árbol BEAST + agregar metadatos por 'label'beast_p <-ggtree(beast_tree, size =1.3) %<+% df_stenopelmatus# Anclar el eje temporal: presente en 0, pasado hacia la izquierda# revts() re-ubica x para que max(x) = 0 (no cambia Y ni rota el árbol)beast_p <-revts(beast_p)# Extraer la tabla interna del plot, contiene coordenadas (-x, y)beast_df <- beast_p$data# Inspeccionar los nombres de columnas disponibles para mapear/visualizarcolnames(beast_df)
Rango de credibilidad bayesiano para la edad de ese nodo bajo el método CA.
CAheight_mean
Edad del nodo (método Common Ancestor, media)
“CA” proviene de Common Ancestor heights (opción de TreeAnnotator). Ajusta edades para ser consistentes entre clados al resumir muchas muestras de árboles.
CAheight_median
Edad del nodo (CA, mediana)
Métrica robusta para reportar edad de clados.
CAheight_range
Rango min–max (CA)
Útil como referencia rápida; prefiere HPD para reportes.
height
Edad del nodo (método estándar de TreeAnnotator)
Si no usaste “CA heights”, esta suele ser la edad principal.
height_0.95_HPD
HPD 95% de la edad (no-CA)
El intervalo de credibilidad más usado en figuras y tablas.
height_median
Edad (mediana)
Igual que arriba, pero explícita la mediana del resumen.
height_range
Rango min–max (no-CA)
Referencia rápida; reporta HPD en manuscritos.
length
Longitud de rama (resumen)
Suele ser la misma magnitud que branch.length pero resumida (media/mediana) por TreeAnnotator sobre el conjunto de árboles.
length_0.95_HPD
HPD 95% de la longitud de rama
Incertidumbre por rama (muy útil con reloj relajado).
length_median
Longitud de rama (mediana)
En árboles datados: tiempo de rama; en no calibrados: sustituciones por sitio.
length_range
Rango min–max de la longitud
Exploratorio.
posterior
Probabilidad posterior del clado
Soporte de clado (0–1). Muy usado para colorear o filtrar nodos.
rate
Tasa molecular de sustituciones por sitio/tiempo.
Solo si usaste reloj relajado: tasa específica de la rama (multiplicador respecto a la media). En reloj estricto, esta columna puede no variar.
rate_0.95_HPD
HPD 95% de la tasa
Incertidumbre de la tasa por rama.
rate_median
Tasa (mediana) por rama
Útil para colorear ramas por tasa estimada.
rate_range
Rango min–max de la tasa
Exploratorio.
🖼️ Ajustar el lienzo: ejes, límites y márgenes
📐 Cómo funcionan los ejes en ggtree
Eje X (x) → representa la distancia acumulada desde la raíz.
Si tu árbol es de Máxima Verosimilitud o inferencia bayesiana, X suele ser sustituciones por sitio.
Si es ultramétrico (ej. BEAST), X puede interpretarse como tiempo.
Eje Y (y) → es un índice que asigna ggtree automáticamente a cada terminal, de arriba a abajo.
No es biológico, solo indica la posición vertical de cada taxón.
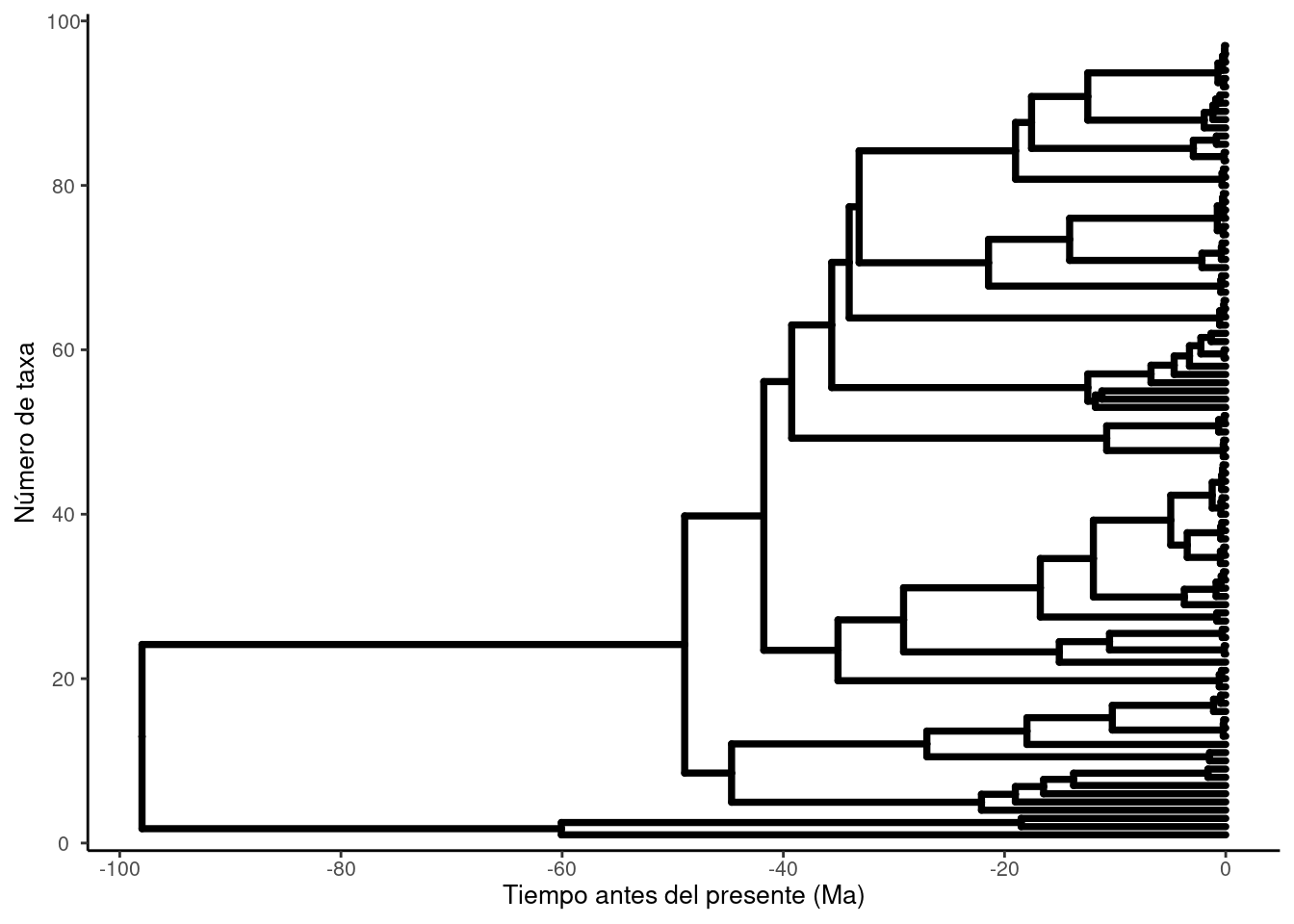
# Rango en X (distancia/tiempo)# xr → te dice de dónde a dónde llega el árbol en horizontal.xr <-range(beast_df$x, na.rm =TRUE)# Rango en Y (posición de tips/nodos)# yr → te da el número mínimo y máximo de posiciones verticales (básicamente cuántos tips).yr <-range(beast_df$y, na.rm =TRUE)# 5% de aire a izquierda/derechapad_left <-0.05*diff(xr)pad_right <-0.05*diff(xr)beast_p +# Eje X visible y ajustadoscale_x_continuous(name ="Tiempo antes del presente (Ma)", # cambia el nombre si es tiempobreaks =pretty(xr, n =6),limits =c(xr[1] - pad_left, xr[2] + pad_right),expand =c(0, 0) # usamos limits, no expand ) +# Eje Y visible con ticksscale_y_continuous(name ="Número de taxa",breaks =pretty(yr, n =5),expand =expansion(mult =c(0.02, 0.04)) ) +# Mostrar ticks y líneas (ggtree los oculta por default)theme(axis.line.y =element_line(),axis.ticks.y =element_line(),axis.text.y =element_text(size =8),axis.title.y =element_text(size =10),axis.line.x =element_line(),axis.ticks.x =element_line(),axis.text.x =element_text(size =8),axis.title.x =element_text(size =10) )
Scale for y is already present.
Adding another scale for y, which will replace the existing scale.
👉 Para que las etiquetas no se corten, el árbol sea legible y la figura mantenga proporciones al exportar.
🔑 Pasos básicos:
📐 Calcular rangos en x y y desde ggtree(tr)$data.
➕ Agregar un poco de aire a la derecha (pad_x) para las etiquetas.
📏 Usar scale_x_continuous y scale_y_continuous con expand=0 para fijar límites exacto
✂️ coord_cartesian(clip="off") → deja que sobresalgan etiquetas largas.
↔︎️ plot.margin → agrega margen extra si se cortan textos.
📊 geom_treescale → coloca una barra de escala en coordenadas reales.
👉 En pocas líneas: controlas el espacio visible, evitas recortes y aseguras consistencia en tus figuras.
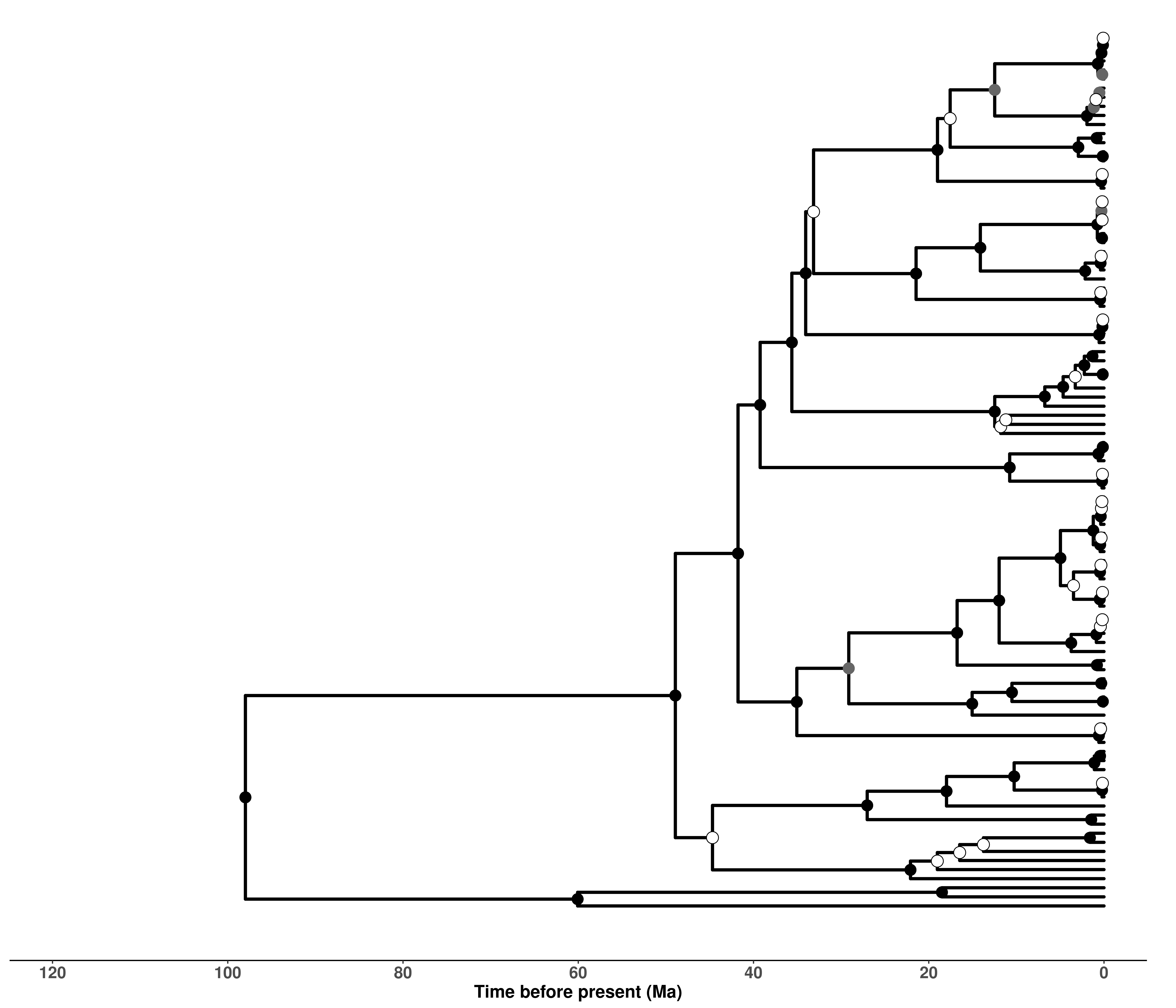
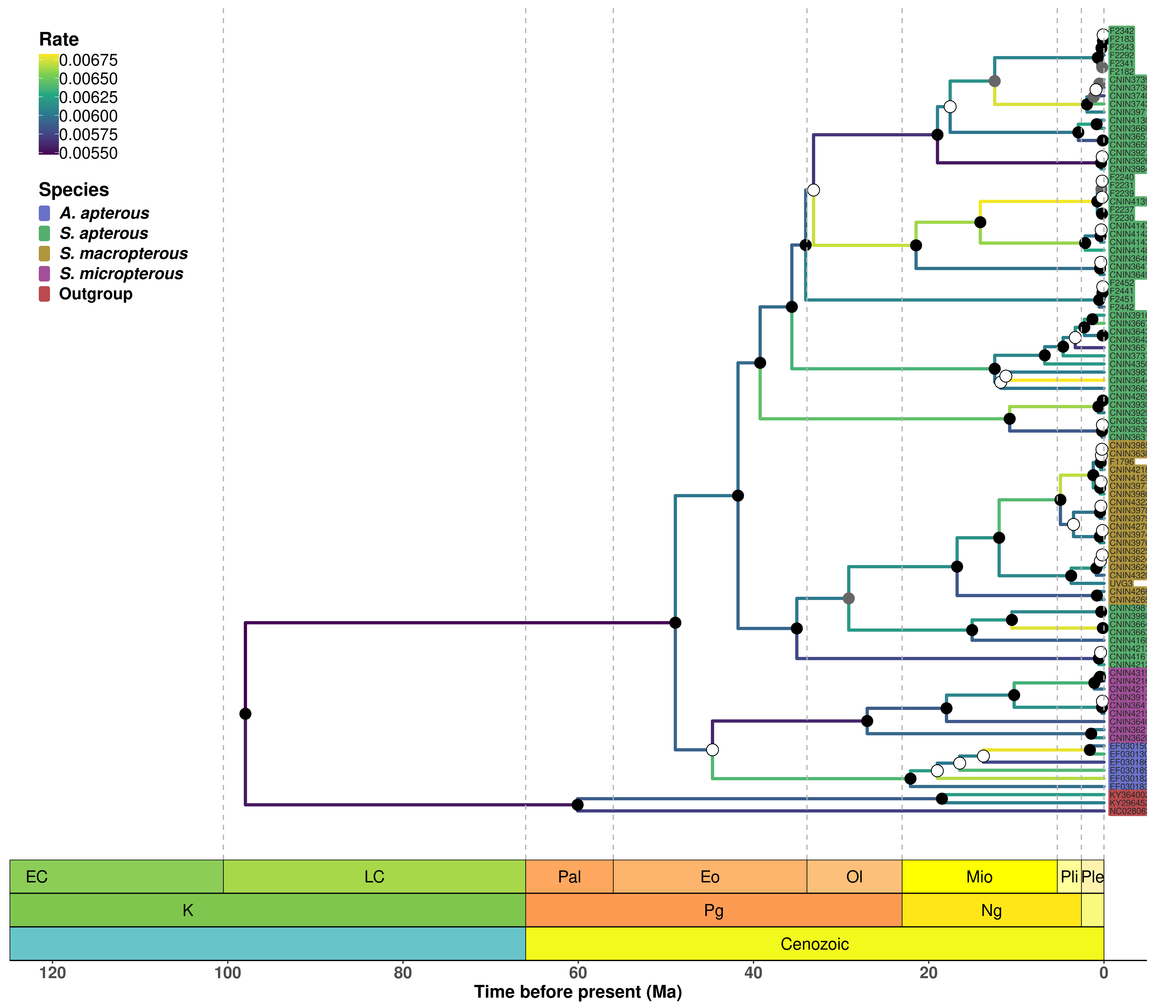
# Extraer información de nodos con edades e intervalos HPD hpd <-as_tibble(beast_tree) %>%select(node, height, height_0.95_HPD) %>%# Nos quedamos con nodo, edad y HPD95 tidyr::unnest_wider(height_0.95_HPD, names_sep ="_") %>%# Desempaqueta la columna list 'height_0.95_HPD' en dos columnas: _1 (mínimo) y _2 (máximo)filter(!is.na(height_0.95_HPD_1), !is.na(height)) # Filtra para quedarnos solo con nodos internos (los tips no tienen HPD)# x: toma el máximo valor del límite superior del HPD x <-max(hpd$height_0.95_HPD_2, na.rm =TRUE)# Redondea "hacia arriba" al múltiplo de 5 más cercano.hdp_max <-ceiling(x /5) *5# obtener el numero de terminalesntips_s <- beast_df %>%filter(isTip =="TRUE") %>%nrow()# Aire extra en X (2% del rango) para no cortar etiquetaspad_x <-0.05*diff(xr)beast_p_adj <- beast_p +scale_x_continuous(name ="Time before present (Ma)",breaks =-seq(0, hdp_max, by =20), # negativos en el datolabels = abs, #pero se ven positivoslimits =c(-hdp_max - pad_x, xr[2] + pad_x),expand =c(0, 0)) +# Limites exactos sin padding automáticoscale_y_continuous(limits =c(-5, ntips_s +3),expand =c(0, 0)) +coord_cartesian(clip ="off") +# Permitir etiquetas largas fuera del paneltheme_tree2() +# dibuja la línea del eje, los ticks y los textos del eje X.theme(plot.margin =margin(5.5, 25, 5.5, 5.5), # Más aire a la derechaaxis.text.x =element_text(size =14, face ="bold"), # tamaño de letra del eje xaxis.title.x =element_text(size =15, face ="bold")) # tamaño de letra del titulo del eje xbeast_p_adj
🛡️Soportes de nodos
En un árbol bayesiano (BEAST, MrBayes), los nodos internos llevan probabilidades posteriores (PP) que indican qué tan confiable es cada clado:
Para figuras más limpias, se puede resumir gráficamente los soportes con un criterio común en filogenia bayesiana de acuerdo a Alfaro y Holder (2006):
👉 Esto permite pasar de una visualización informativa (números) a una visualización clara y estética basada en categorías de soporte.
# Puntos por umbral de PP (0–1)beast_p_adj <- beast_p_adj +# Alta: PP ≥ 0.95 → negrogeom_point2(aes(subset =!isTip &!is.na(posterior) & posterior >=0.95),size =4.5, shape =16, color ="black") +# Moderada: 0.90–0.95 → grisgeom_point2(aes(subset =!isTip &!is.na(posterior) & posterior >=0.90& posterior <0.95),size =4.5, shape =16, color ="grey40") +# Baja: PP < 0.90 → blancogeom_point2(aes(subset =!isTip &!is.na(posterior) & posterior <0.90),size =4.5, shape =21, fill ="white", color ="black")beast_p_adj
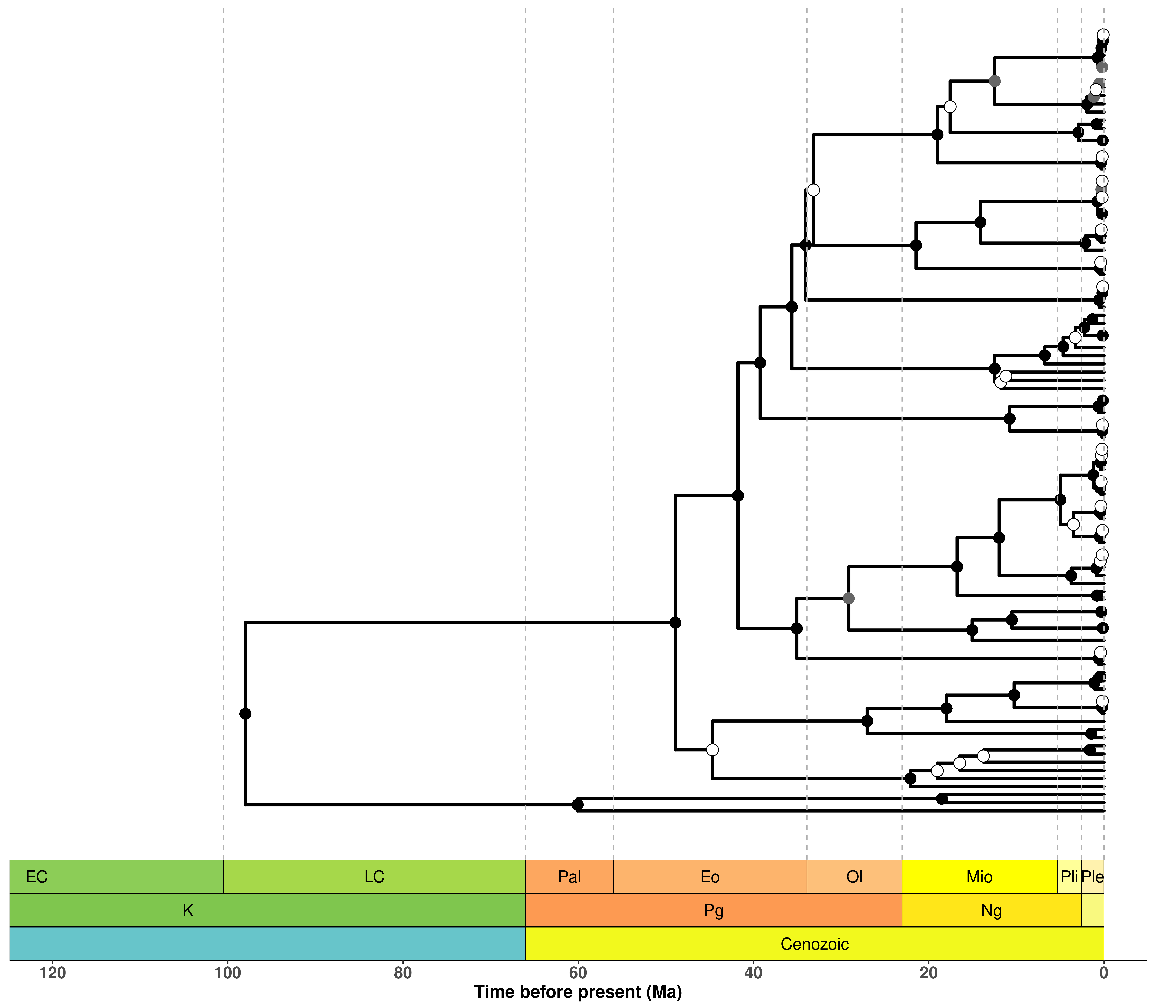
🕰️ Árbol datado con escala temporal y referencia estratigráfica
Para facilitar la lectura cronológica del árbol, añadimos dos elementos:
Faja geológica al pie del gráfico (eras, períodos y epochs) usando deeptime::coord_geo(). Esto da un contexto estratigráfico claro para interpretar las edades de divergencia.
Líneas verticales punteadas en los límites de los epochs, alineadas con el eje temporal (0 en el presente y valores negativos hacia el pasado).
Estas guías ayudan a ubicar rápidamente cada nodo respecto a cambios geológicos mayores.
# Límites de epochs (Ma) en eje negativo --------------------------------# deeptime::epochs trae min_age/max_age (Ma). Convertimos a eje negativo (pasado < 0).epochs_df <- deeptime::epochs %>%select(min_age, max_age)epoch_limits <-sort(unique(c(-epochs_df$min_age, -epochs_df$max_age, 0)))beast_p_adj <- beast_p_adj +geom_vline(xintercept = epoch_limits,linetype ="dashed", linewidth =0.5, color ="grey70") +coord_geo(dat =list("epochs", "periods", "eras"), # tres nivelespos =list("b", "b", "b"), # colocar en la parte inferiorneg =TRUE, # tiempo negativo hacia el pasadoabbrv =list(TRUE, TRUE, FALSE) # abreviados: epochs/periods; eras completas )beast_p_adj
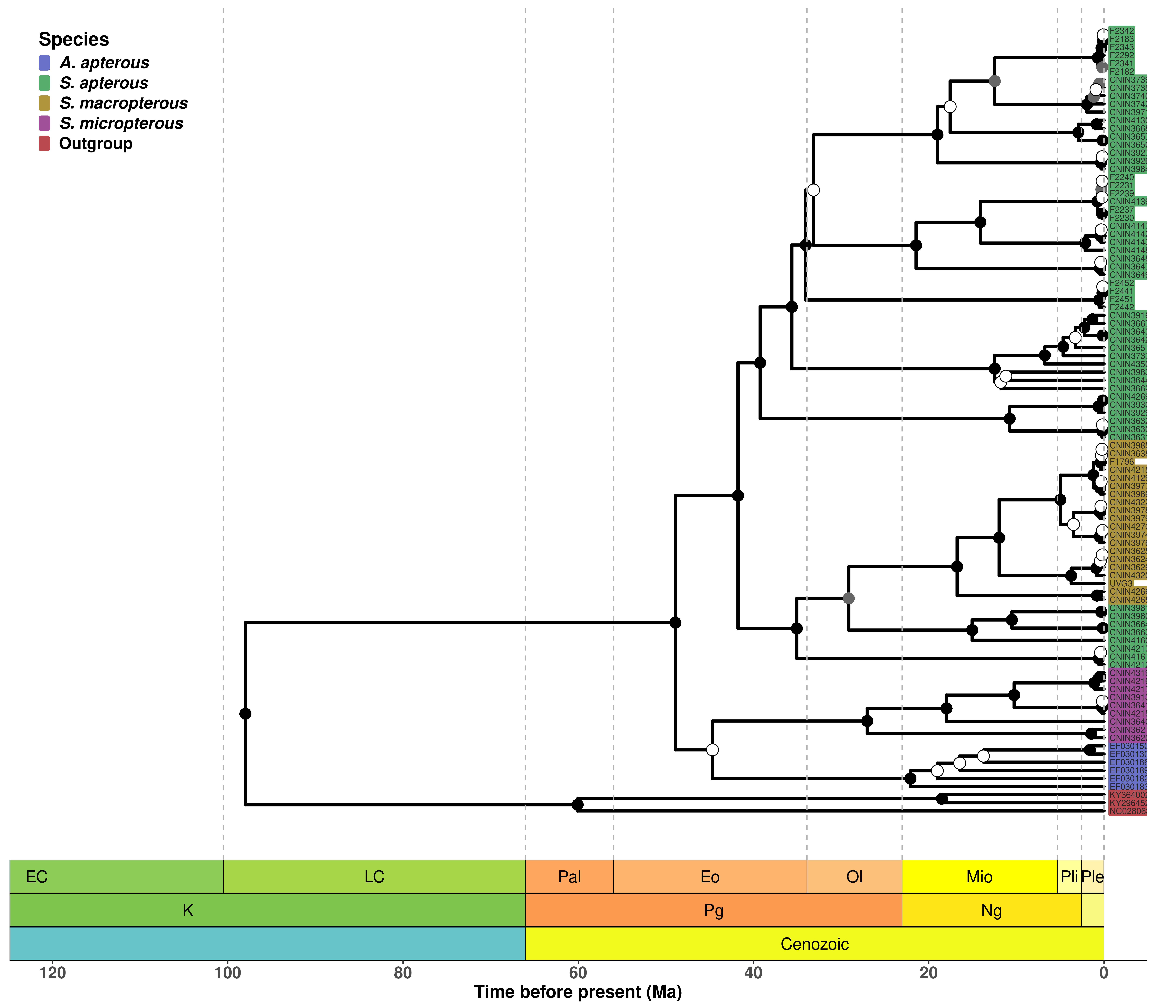
🎨 Etiquetas de las terminales
En esta parte vamos a enriquecerlo con nuestros metadatos —que añadimos en el paso previo con el operador %<+% de ggtree— para poder representar información extra en el árbol.
🔤 Pintar los nombres de las terminales por categorías (ej. S. apterous, S. micropterous…).
# Paleta por categoría funcional (ajústala si quieres)pal <-c("Stenopelmatus_apterous"="#56ae6c","Stenopelmatus_micropterous"="#a24f99","Stenopelmatus_macropterous"="#af953c","Ammopelmatus_apterous"="#6971c9","Outgroup"="#ba4a4f")beast_p_adj <- beast_p_adj +geom_tiplab(aes(fill = Species, label = label), # fill por especiecolor ="grey15",geom ="label", # cajita detrás del textolabel.size =0, # sin borde en la cajitalabel.padding =unit(0.12, "lines"), # relleno interno de la cajitasize =2.6,offset =0.003,show.legend =TRUE ) +scale_fill_manual(values = pal,na.value ="black",# orden explícito de los gruposbreaks =c("Ammopelmatus_apterous","Stenopelmatus_apterous", "Stenopelmatus_macropterous","Stenopelmatus_micropterous","Outgroup"),labels =c(Ammopelmatus_apterous =expression(bolditalic("A. apterous")),Stenopelmatus_apterous =expression(bolditalic("S. apterous")),Stenopelmatus_macropterous =expression(bolditalic("S. macropterous")),Stenopelmatus_micropterous =expression(bolditalic("S. micropterous")),Outgroup =expression(bold("Outgroup")))) +guides(fill =guide_legend(title ="Species",# símbolo cuadrado, sin letra y un poco más pequeño para que el gap sea menoroverride.aes =list(shape =15, size =4, label =NULL, colour =NA, stroke =0),label.hjust =0, title.hjust =0)) +theme(legend.position =c(0.02, 0.98),legend.justification =c("left", "top"),legend.background =element_rect(fill ="white", color ="white"),# menos espacio entre key y textolegend.title =element_text(size =16, face ="bold"),legend.text =element_text(size =14, margin =margin(l =0)) )beast_p_adj
🌡️ Colorear ramas por la tasa molecular
En un análisis de BEAST con reloj relajado, cada rama puede tener una tasa de sustitución distinta (subs/sitio/tiempo).
Colorear el árbol con la paleta viridis permite visualizar qué linajes evolucionan más lento (azules/oscuros) o más rápido (amarillos/claros) a nivel molecular, resaltando la heterogeneidad en la velocidad de acumulación de mutaciones.
# Rango de tasas sin considerar el valor de la raíz = 1 rate_rng <-range(beast_p$data$rate[beast_p$data$rate !=1], na.rm =TRUE)# Árbol con ramas coloreadas por tasa molecularbeast_p_rate <- beast_p_adj +aes(color = rate) +# asigna el color al valor de 'rate' por ramascale_color_viridis_c(limits = rate_rng, # recorta la escala ignorando la raíz = 1 na.value ="grey80", # color neutro para valores faltantesname ="Rate") # título de la leyenda# Visualizarbeast_p_rate
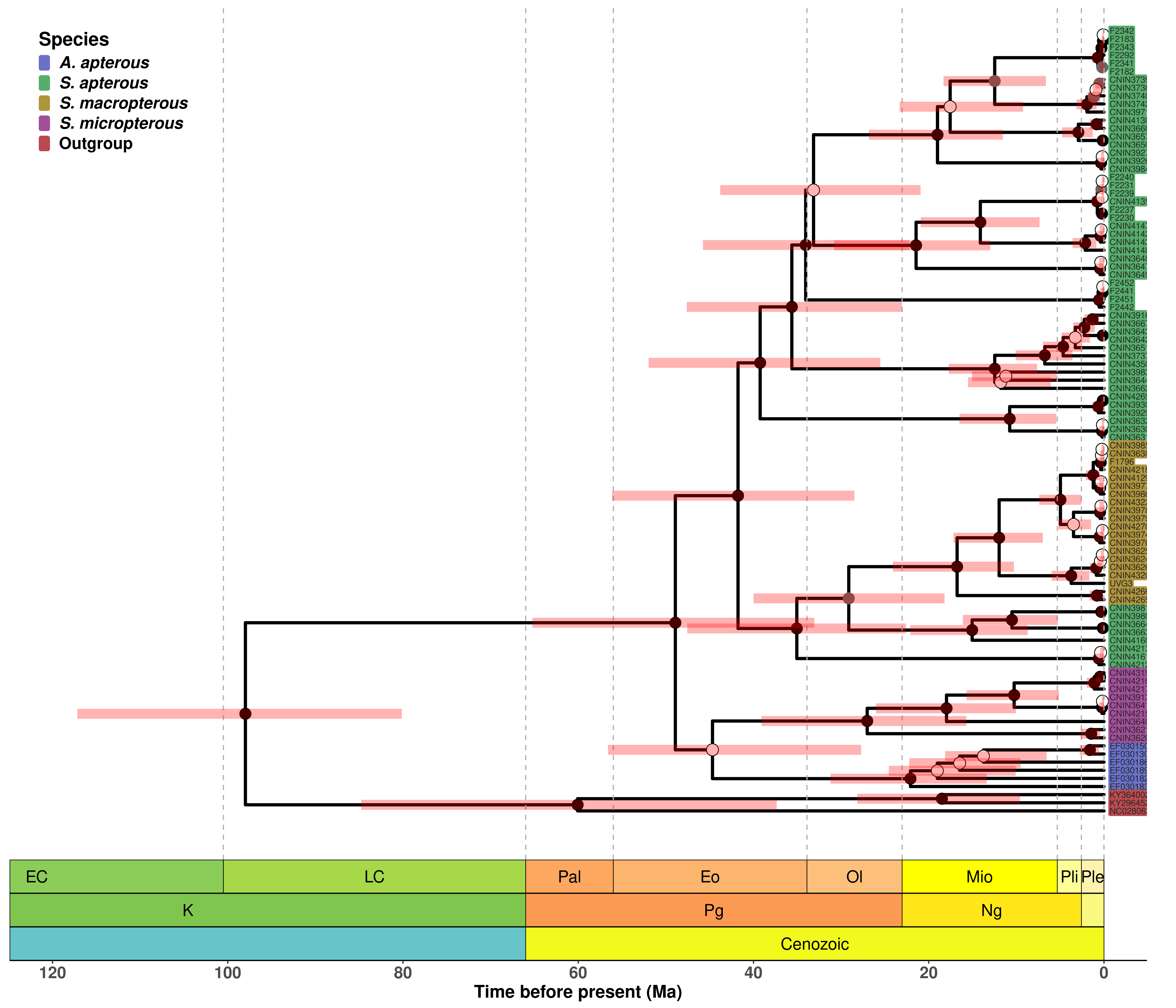
📊 Barras de incertidumbre (HPD) en árboles datados
En los árboles datados obtenidos con BEAST, además de las edades estimadas de los nodos, es fundamental mostrar la incertidumbre asociada.
Las barras HPD (Highest Posterior Density) al 95% representan el rango de edades más creíble para cada divergencia según la inferencia bayesiana.
Visualizar estas barras junto con la filogenia:
Da una idea clara de la precisión temporal de las estimaciones.
Permite identificar clados con mayor incertidumbre en comparación con otros.
Es una práctica estándar en publicaciones, ya que comunica tanto el valor puntual (mediana) como el intervalo de credibilidad.
# Extraer coordenadas Y de cada nodo a partir del objeto gráfico ---------------coords <- beast_p_adj %>%select(node, y) # Cada nodo tiene asignada una coordenada 'y' en la gráfica# Unir coordenadas Y con las edades HPD de cada nodo ---------------------------hpddf <-left_join(coords, hpd, by ="node")# Dibujar barras horizontales representando los intervalos HPD -----------------beast_p_adj +geom_segment(data = hpddf,aes(x = height_0.95_HPD_1 *-1, # Límite inferior de la barra (invertido a negativo para el eje tiempo)xend = height_0.95_HPD_2 *-1, # Límite superior de la barray = y, yend = y), # Mantiene la barra horizontal a la altura del nodolinewidth =4, # Grosor de la barraalpha =0.3, # Transparenciacolor ="red"# Color de las barras HPD )
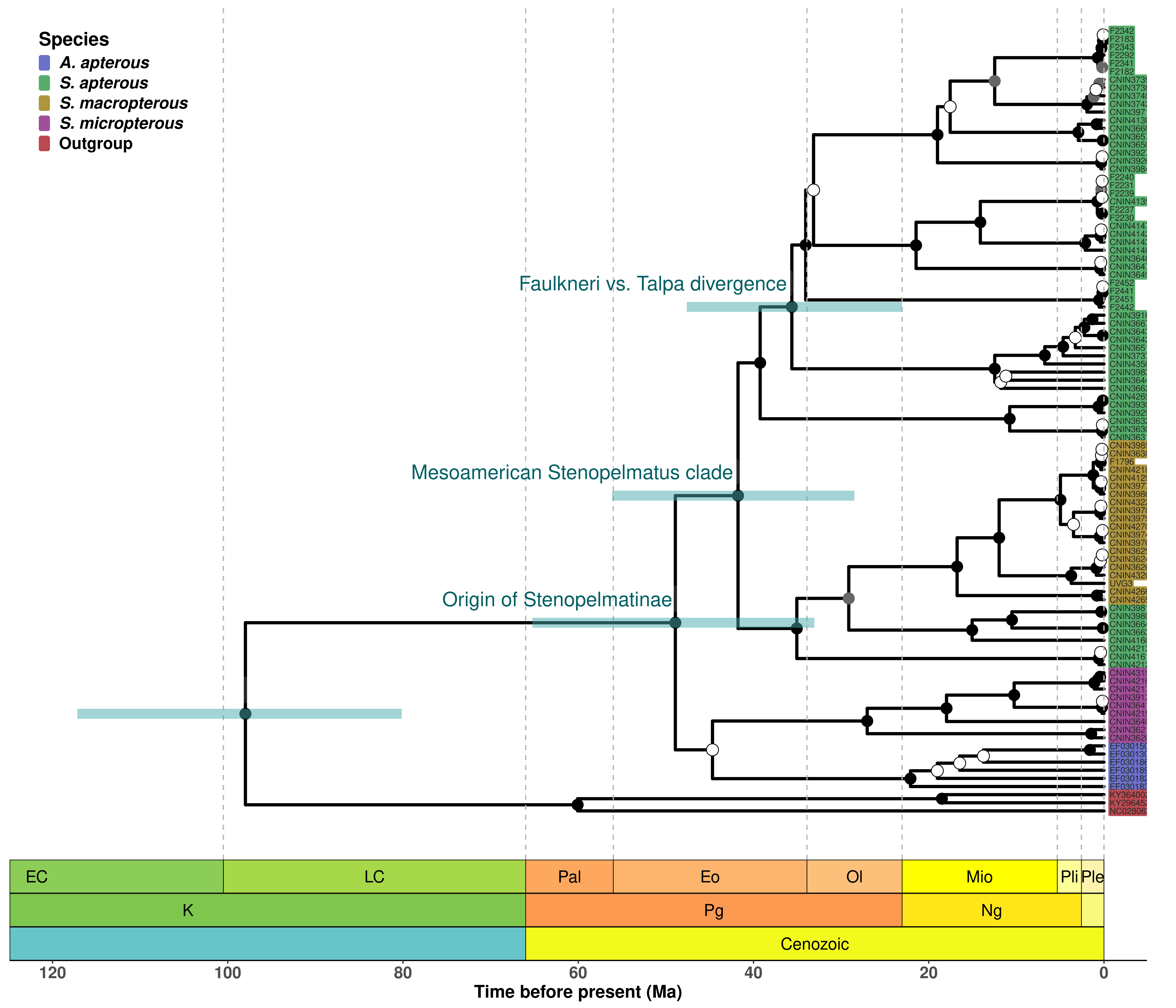
🎯 Barras HPD en nodos clave
En algunos casos, mostrar todas las barras HPD del árbol puede generar saturación visual y dificultar la interpretación.
Una alternativa útil es resaltar únicamente los nodos de interés biológico o evolutivo, como los que marcan orígenes de clados principales.
👉 Aquí destacamos cuatro nodos clave en la historia de Stenopelmatinae, mostrando sus intervalos de credibilidad (HPD 95%) y la mediana de sus edades estimadas: