Introducción
¿Qué es un árbol filogenético?
Un árbol filogenético es una representación gráfica que describe las relaciones evolutivas entre un conjunto de organismos o secuencias, basada en datos moleculares, morfológicos o en ambos.
🔎 Importante:
- Es un modelo de hipótesis → no representa la verdad absoluta, sino una inferencia construida a partir de datos y métodos analíticos.
🌿 ¿Qué representan los árboles filogenéticos?
🔗 Relaciones evolutivas: cómo están relacionadas las especies entre sí.
📏 Distancia genética: medida de cambio evolutivo (por ejemplo, el número de sustituciones por sitio).
⏳ Tiempo evolutivo: en el caso de árboles ultramétricos.
🌳 Eventos de especiación: reflejados en los nodos internos.
💡 Nota: en árboles obtenidos con métodos de distancia (filogramas de similitud), el enfoque es distinto: representan “quién se parece más a quién”, pero no reconstruyen ancestros ni relaciones evolutivas directas.
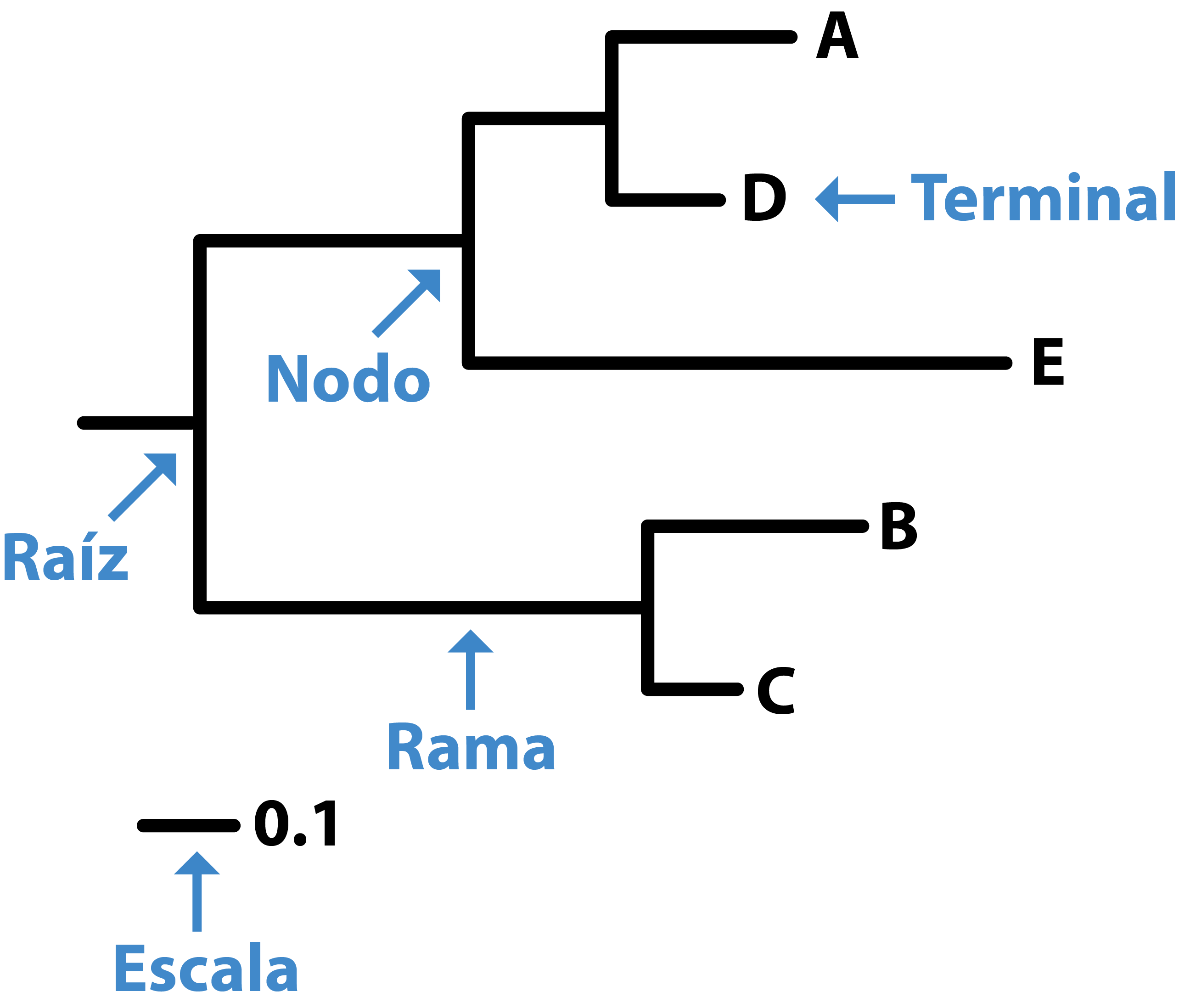
🧩 Componentes del árbol
🌳 Raíz: nodo que representa el ancestro común más antiguo.
🔘 Nodo: punto donde se bifurca el árbol, representando un ancestro común hipotético.
🏷️ Terminal (tip): representa una especie, gen o secuencia observada.
➖ Rama: línea que conecta nodos; puede estar escalada según distancia genética o tiempo.
📏 Escala: barra de referencia que indica cuánto equivale una unidad de longitud de rama.
En árboles de distancia genética, número de sustituciones (cambios) esperados por sitio.
En árboles ultramétricos, la escala corresponde a tiempo evolutivo (millones de años, por ejemplo).
🌲 Tipos de árboles filogenéticos
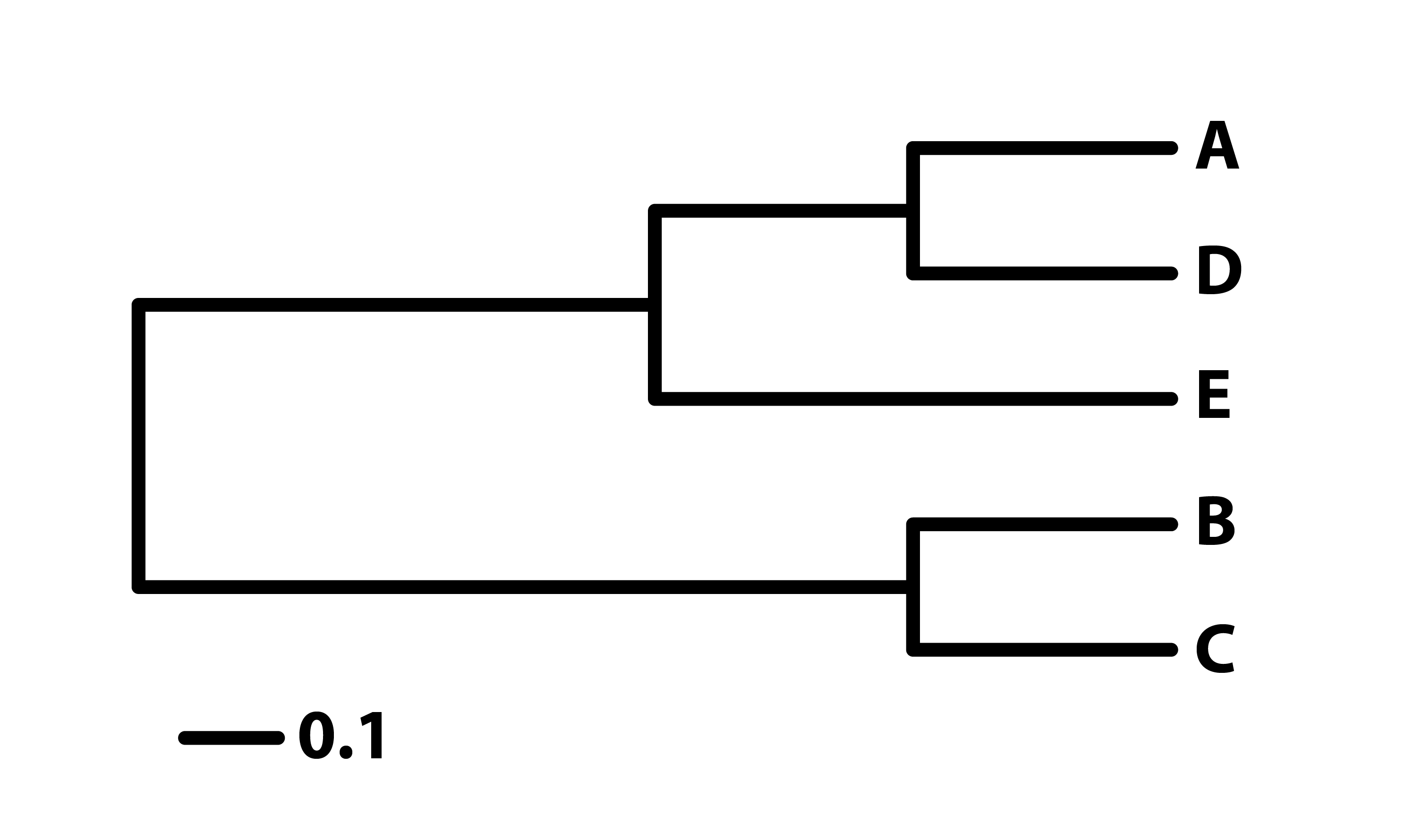
🌳 Enraizado
Cuenta con una raíz que marca el ancestro común más antiguo y define la dirección del tiempo evolutivo.
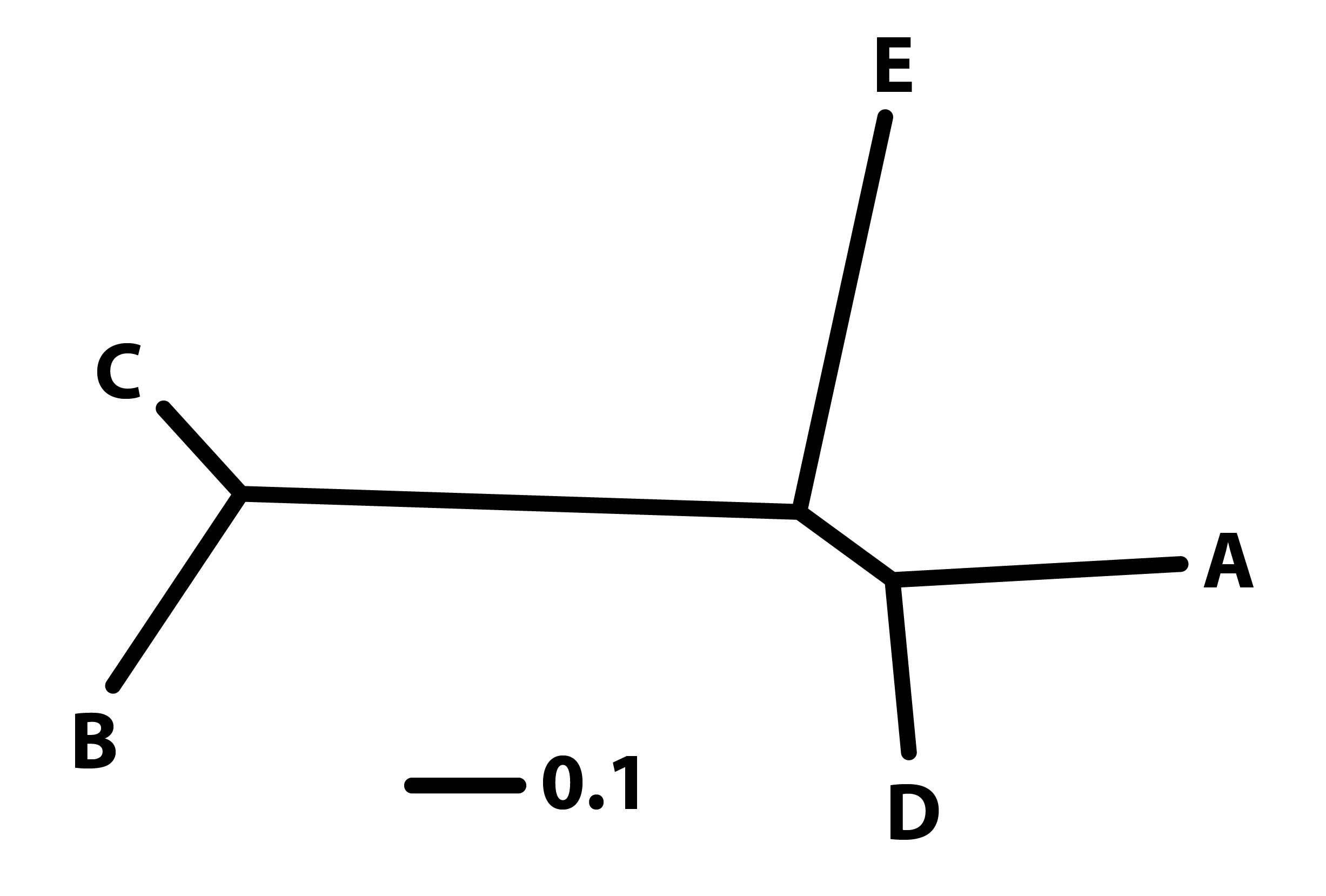
🌿 No enraizado
Muestra las relaciones entre los taxa, pero no indica la dirección temporal de la evolución.
⏳ Ultramétrico
Todos los terminales están a la misma distancia de la raíz. Este tipo de árbol se obtiene, por ejemplo, al aplicar relojes moleculares para estimar tiempos de divergencia.
📏 Escalado por distancia
La longitud de las ramas es proporcional a la cantidad de cambio evolutivo, como el número de sustituciones por sitio en secuencias moleculares.
🧪 Tipos de métodos para construir árboles
🧩 A) Métodos basados en distancias
Calculan una matriz de distancias genéticas y construyen un árbol que mejor las represente.
📊 Ejemplos: Neighbor-Joining (NJ), UPGMA.
💻 Programas: MEGA, PHYLIP.
🪶 B) Métodos de parsimonia
Buscan el árbol que minimiza el número total de cambios evolutivos.
⚡ Más simples y rápidos, pero pueden ser inconsistentes cuando los datos tienen alta homoplasia.
💻 Programas: PAUP*, TNT, MEGA.
🎲 C) Métodos probabilísticos
🔹 Máxima Verosimilitud (ML)
Encuentra el árbol que maximiza la probabilidad de observar los datos bajo un modelo de evolución.
- 💻 Programas: IQ-TREE, RAxML, PhyML.
🔹 Inferencia Bayesiana
Estima la probabilidad posterior de los árboles dado los datos y un modelo de evolución, usando MCMC para explorar el espacio de árboles.
- 💻 Programas: MrBayes, BEAST, RevBayes.
📂 Formatos y salidas de programas
| Programa | Extensión típica | Formato | Árbol ultramétrico | Comentarios |
|---|---|---|---|---|
| RAxML / RAxML-NG | .tree, .tre, .bestTree |
Newick | ❌ No (por defecto) | Incluye longitudes de rama en sustituciones por sitio; usa ML. |
| MrBayes | .t, .tre |
Nexus | ❌ No (a menos que se use reloj molecular) | Guarda múltiples árboles de la cadena MCMC; puede exportar a Newick. |
| IQ-TREE | .treefile |
Newick | ❌ No (por defecto) | Archivo principal con longitudes de rama y soporte de nodos. |
| BEAST | .trees |
Nexus o Newick | ✅ Sí (por defecto) | Produce árboles calibrados en tiempo; requiere reloj molecular. |
| RevBayes | .trees, .tree |
Nexus o Newick | ⚖️ Depende del modelo | Muy flexible; formato definido por el usuario. |
| PAUP* | .tre |
Nexus o Newick | ⚖️ Depende del análisis | Compatible con parsimonia, ML y distancias. |
| MEGA | .nwk, .meg |
Newick | ⚖️ Depende del análisis | Interfaz gráfica; exporta fácilmente a Newick. |
| PHYLIP | .tree, .tre |
Newick | ⚖️ Depende del programa interno | Salida muy básica; normalmente Newick sin etiquetas complejas. |
📑 Formato Newick y Nexus
Existen varios formatos para almacenar árboles, pero los más comunes son Newick y Nexus. Casi todos los programas utilizan alguno de estos dos, aunque con diferentes extensiones de archivo (.tree, .tre, .t, .treefile, etc.).
🌿 Newick
El formato más utilizado y universal. Es simple y compacto:
((A:0.1,B:0.2):0.3,C:0.4); 🔹 Características:
Usa paréntesis
( )para representar la topología (quién es hermana de quién).Las longitudes de rama se indican con
:valor.Siempre termina con
;.Puede incluir información extra como soporte o probabilidades en etiquetas o comentarios.
🔹 Ejemplos de anotaciones:
Soporte como etiqueta clásica:
(A,B)90:0.05;Soporte como metadato explícito:
(A,B)[&pp=0.98]:0.05;
📦 Nexus
El formato más flexible, usado sobre todo en análisis bayesianos y programas que requieren guardar metadatos.
Ejemplo mínimo:
#NEXUS Begin trees; Tree myTree = ((A:0.1,B:0.2):0.3,C:0.4); End; 🔹 Características:
Comienza con
#NEXUS.Organiza la información en bloques (
Begin … End;).Permite guardar múltiples árboles en un mismo archivo.
Puede incluir particiones, modelos de evolución, soporte, tiempos, HPD, etc.
🌿 Formato Newick: cómo leerlo y escribirlo
El formato Newick es el estándar más extendido para representar árboles filogenéticos en texto plano. Es compacto, portable y lo admiten prácticamente todos los programas.
📏 Reglas de sintaxis esenciales en Newick
Paréntesis = clados
Cada par de paréntesis define un clado.
👉 Ejemplo:(A,B)Significa que A y B son hermanas.
Comas = separación de taxones o clados
Los elementos dentro de un mismo nivel se separan con comas.
👉 Ejemplo:((A,B),C)
Aquí C es hermana del clado (A,B).
Longitudes de rama =
:valor
Las longitudes (ej. sustituciones por sitio) se colocan después de cada taxón o nodo.
👉 Ejemplo:(A:0.1,B:0.2):0.05A → rama de longitud 0.1
B → rama de longitud 0.2
Nodo interno (A,B) → rama de longitud 0.05
Todo árbol termina con
👉 Ejemplo:((A,B),C);Soportes en nodos internos
👉 Ejemplo:(A,B)90:0.05
💡 Los soportes (90, 85) se colocan después del ) del clado y antes de :longitud de la rama interna de ese clado.